


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Keith Alan DeHaas, MLS(ASCP)CM⇑
- Address for Correspondence: Keith Alan DeHaas, MLS(ASCP)CM, 148 Convent Avenue, Bennington, VT, 05201, 802-681-8760, keith.dehaas{at}gmail.com
Abstract
Traditionally, anticoagulant therapy has been conducted through the administration of vitamin K antagonists (VKAs) and parenteral alternatives. Direct Oral Anticoagulants (DOACs) such as apixaban, rivaroxaban and edoxaban provide the convenience of the VKA in that they are administered orally yet they lack many of the disadvantages associated with VKA therapy. Unlike VKAs, measuring DOAC levels is usually unnecessary. However, there are instances with plasma concentrations should be determined. The chromogenic anti-Xa assay is the best linear quantitative assay that is easily accessible to most clinical laboratories for this purpose. However, since the calibrators and controls for apixaban, rivaroxaban and edoxaban are awaiting Federal Drug Administration (FDA) clearance, measurement may have to be performed using the Prothrombin Time assay that lacks the sensitivity of the chromogenic anti-Xa. Results should be periodically verified using liquid chromatography in tandem with mass spectrometry which is the reference method for measuring DOAC levels in plasma.
ABBREVIATIONS: AF-atrial fibrillation, DOAC – direct oral anticoagulant, DVT – deep vein thrombosis, FDA – Food and Drug Administration, HPLC – high performance liquid chromatography, LMWH – low molecular weight heparin, PE – pulmonary embolism, PT – prothrombin time, UFH – unfractionated heparin, UPLC – ultra-performance liquid chromatography, VKA – vitamin K antagonist, VTE – venous thromboembolism
INTRODUCTION
Anticoagulant agents are employed to reduce the risk of complications caused by thromboembolic disease. Thromboembolic disease may be arterial or venous1 such as venous thromboembolism (VTE). This condition is caused by an imbalance of clotting factors and endogenous anticoagulants, which leads to the formation of a thrombus.2 It includes both deep vein thrombosis (DVT) and pulmonary embolism (PE) and can often be life threatening. VTE results from a combination of hereditary and acquired risk factors. Hospitalization is responsible for nearly 66% of all VTE events and approximately 300,000 of these patients die.3 Pulmonary embolism is the third most common cause of hospital-related death and the most common preventable cause of hospital-related death. Surgery and especially orthopedic surgery continues to be significant risk factor for VTE. In fact, after major elective orthopedic surgery the incidence of a VTE is estimated to be 40% - 60% if anticoagulant therapy is not applied.1 Conditions such as cardiac arrhythmias also require anticoagulant intervention. Atrial fibrillation (AF) is the most common cardiac arrhythmia. It is found in one percent of the population and in nine percent of people over the age of 80 years and is a risk factor for stroke. An individual with this condition is 5 times more likely to suffer a stroke than a person who lacks this condition.4 The chance of men and women over the age of 40 developing AF is 25%.2
Historically, vitamin K antagonists (VKAs) such as warfarin (Coumadin) have formed the backbone of anticoagulant therapies for treatment of thromboembolic disease and cardiac arrhythmias. VKAs function as an anticoagulant by altering the hepatic synthesis of multiple clotting proteins and are traditionally used for long term anticoagulant treatment. Parenteral agents such as unfractionated heparin (UFH), low molecular weight heparin (LMWH) and fondaparinux are generally used to initiate an immediate anticoagulant response. Heparin acts on multiple proteases in the clotting cascade, whereas fondaparinux is an indirect factor Xa inhibitor. The benefit of VKAs over parenteral therapies is that they can be applied orally and are less expensive. The disadvantage is that they cross react with many medications and foods, take 36 – 72 hours to reach their very narrow therapeutic range, have a wide range of pharmocodynamic responses at a given blood concentration across individual patients. For this reason they require constant monitoring and dose adjustments.1 The disadvantage of parenteral agents is their expense and their inability to be given orally. This prevents them from being a long term solution, however because they act quickly they can be used as an anticoagulant bridge until the slower acting VKA can take effect.5 Despite their many limitations, there is a great deal of evidence supporting the use of VKAs and parenteral agents and they are recommended by the American College of Chest Physicians and the European Society of Cardiology for anticoagulant therapy.1
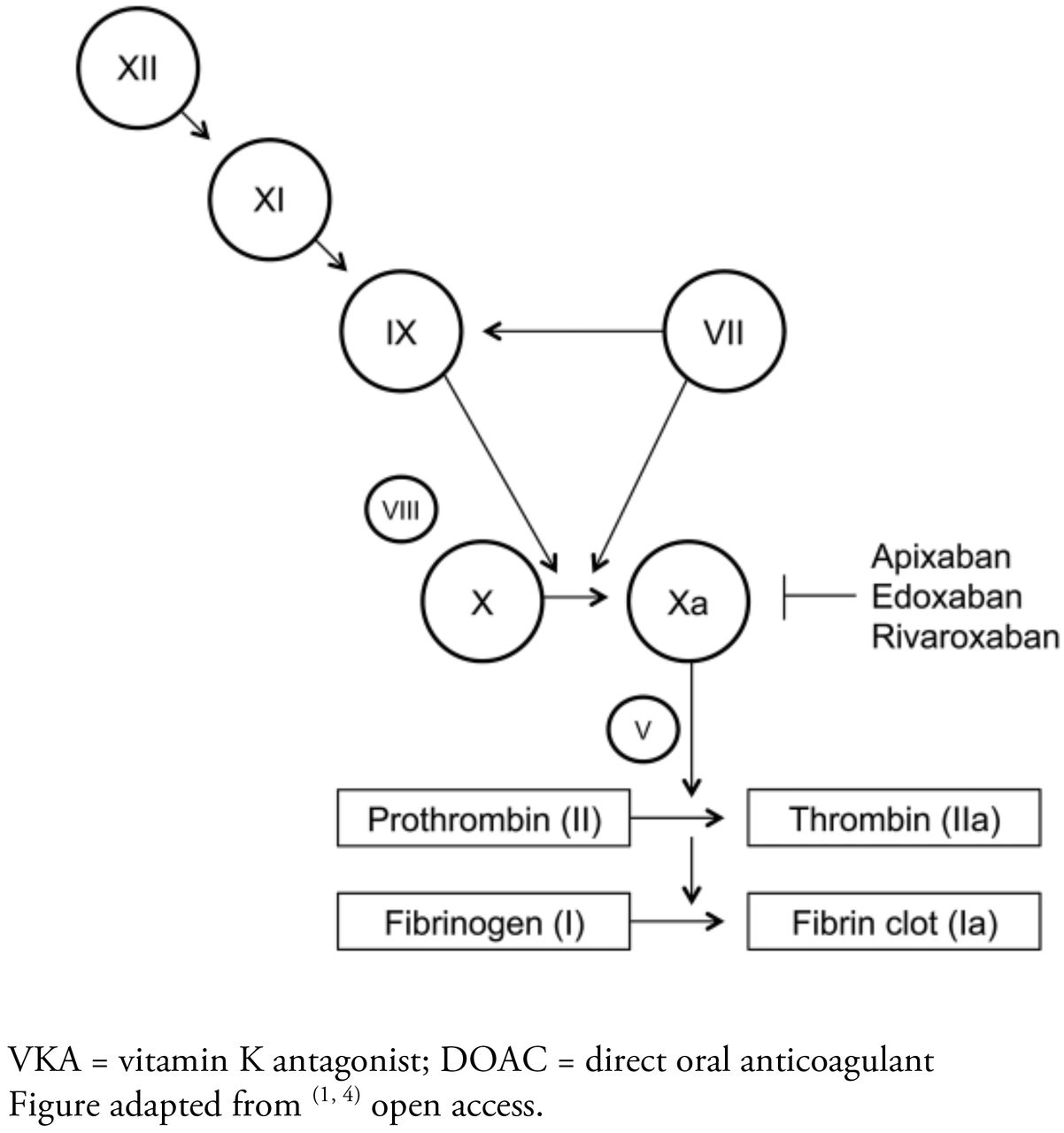
A major shift from VKA anticoagulant therapies was introduced through the development of direct oral anticoagulants (DOACs) for the treatment of thromboembolic disease. The DOACs apixaban (Eliquis), rivaroxaban (Xarelto) and edoxaban (Savayasa, Lixiana) have undergone large phase III clinical trials for various thromboembolic and cardiovascular conditions. All three drugs have Food and Drug Administration (FDA) approval for the treatment of thromboembolic disorders and prevention of ischemic stroke in cardiac arrhythmias.3,6,7 Like VKAs, DOACs are administered orally, but unlike VKAs where the mode of action involves multiple clotting components within the cascade, DOACs have a very specific target (see Figure 1). They achieve their anticoagulant effect by stoichiometrically inhibiting factor Xa (the DOAC dabigatran inhibits thrombin but it is not being discussed in this review). DOACs also have more predictable pharmocodynamic properties and fewer food and drug interactions than VKAs. Finally, DOACs can reach their full therapeutic effect within 4 hours and have half-lives of less than 24 hours. Despite these advantages, many providers continue to use VKAs for long term management because they have little clinical experience with DOACs and VKAs have represented the gold standard for thromboembolic therapy for a long time.1

Target Specificity of DOACs Compared to VKAs
Mechanism of Action
Apixaban is a direct factor Xa inhibitor. It does not require antithrombin for antithrombotic activity. Apixaban inhibits free and clot-bound factor Xa and prothrombinase activity. Apixaban has no direct effect on platelet aggregation but indirectly inhibits platelet aggregation induced by thrombin. By inhibiting factor Xa, apixaban decreases thrombin generation and thrombus development. Apixaban has greater than 50% bioavailability and reaches peak plasma concentration in 3 to 4 hours and the terminal half-life is 10 to 14 hours after repeated doses. It is partially metabolized by CYP3A4, the most common and versatile member of the P450 group of oxidizing enzymes. Approximately 25% is eliminated through the kidneys and to some degree also processed via CYP-independent mechanisms in the liver. Because it does not inhibit or induce CYP enzymes, it is expected to have a low likelihood of drug to drug interactions.5
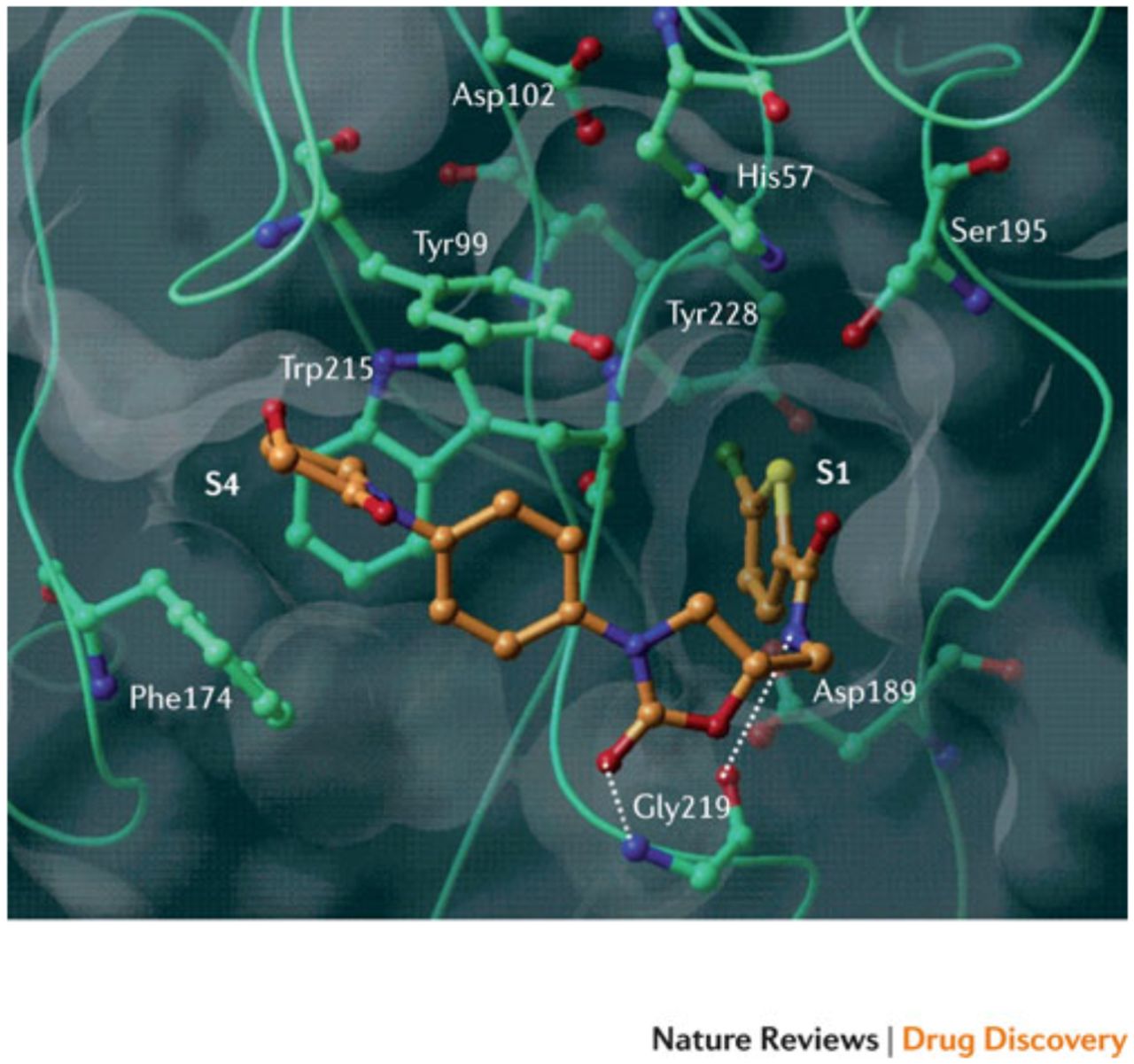
Rivaroxaban was the first oral, direct factor Xa inhibitor. It is an oxazolidinone derivative that binds directly and reversibly to factor Xa via the S1 and S4 pockets (see Figure 2). Rivaroxaban competitively inhibits factor Xa and is more than 10,000-fold more selective for factor Xa than other related serine proteases, and it does not require cofactors (such as antithrombin) to exert its anticoagulant effect. Unlike indirect factor Xa inhibitors, rivaroxaban inhibits both free and clot-bound factor Xa, as well as prothrombinase activity, thereby prolonging clotting times.8 When taken orally, the 10 mg dose has 80 – 100% bioavailability regardless of food intake, but the 10 mg, 15 mg and 20 mg dose bioavailability appears to be dose proportional. Rivaroxaban has a half-life of 5 – 9 hours in the plasma of the young and within 11 – 13 hours in the elderly. About 2/3 of an administered dose undergoes metabolic degradation of which 50% is eliminated via the hepatobiliary route and 50% renally. The remaining 1/3 is eliminated renally through the urine as active substance without being altered in any way.9
Figure 2 shows the X-ray crystal structure of a DOAC (rivaroxaban) in complex with human factor Xa. Essential amino acids and binding pockets (S1 and S4) are indicated; hydrogen bonds are shown as dotted lines. Illustration adapted with permission of publisher.9

Sites of Interaction between DOAC and factor Xa
Like apixaban and rivaroxaban, edoxaban is a highly specific direct inhibitor of factor Xa. It is rapidly absorbed and its pharmacokinetics and pharmacodynamics do not seem to be affected by food intake. After absorption into the blood, the majority of the edoxaban dose remains unaltered. However, several metabolites do occur mostly through hydrolysis. A few metabolites are also produced via cytochrome P4503A enzymes. Although peak plasma concentration and other pharmacodynamic characteristics differ among the formulations, bioavailability is similar between the tablet and solution forms of the drug. Peak concentrations are reached one to two hours post dose. Edoxaban spreads into extravascular tissues with little protein uptake (40% to 59%) which remains constant up to 12 hours post dose. The half-life ranges from 5.8 to 10.7 hours for both the tablet and solution formulations (in single- and multiple-dose regimens). Edoxaban is eliminated renally at a rate exceeding the glomerular filtration rate indicating active secretion into the kidney. Approximately 60% of edoxaban is eliminated in the feces and 40% through the urine.2
Overview of Selected Oral Anticoagulant Agents in the U.S.
Laboratory analysis
The laboratory monitoring of DOACs is not recommended due to the predictable nature of their pharmocodynamic and pharmacokinetic properties. However, there are instances when it might be desirable to ascertain the activity levels of the anticoagulant. These instances may include recurrent thrombosis, bleeding events, an urgent surgery, to estimate patient compliance or suspected overdose and when bridging with other anticoagulants.7,10 A phase II trial involving patients with non-valvular AF indicated that the tendency to bleed increased with the size of the administered dose of edoxaban. Also, a subanalysis of patients included in the ENGAGE-AF TIMI 48 study showed high variability in plasma peak and trough concentrations with the 60mg dose. This is concerning since the data points to bleeding and stroke events correlating closely with the plasma concentrations at trough. Therefore, monitoring may be useful in ensuring safe use of edoxaban in frail patients.3
The best laboratory test with which to obtain accurate measurements of DOAC concentration in plasma is the linear and quantitative chromogenic anti-Xa assays, using calibrators specific for the particular DOAC being measured.7,10 Apixaban and rivaroxaban calibrators are commercially available for this purpose. However, edoxaban calibrators are not available as of this writing so an anti-Xa assay specifically dedicated for the measurement of direct factor Xa inhibitors should be used for this DOAC. Normal patient plasma samples spiked with different concentrations of edoxaban can be used as calibrators. Specific chromogenic anti-Xa assays developed for the quantitative measurement of direct FXa inhibitors are the most practical and dependable assays for the evaluation of edoxaban concentrations in plasma.3
Chromogenic anti-Xa assays are less sensitive than clotting tests to sample collection conditions and patient to patient variability pertaining to the amounts of clotting factors present. This allows for a reduction in variability of testing between individual patients. The sensitivity of these assays is affected by the methodologies, the chromogenic substrate, the exogenous factor Xa and the ratio between these reagents. In addition, factors such as the ionic force and the pH of the buffer solution could also affect the sensitivity of a particular assay. Therefore, confirmation of the accuracy of these chromogenic anti-Xa assays using specific calibrators and controls should be compared to liquid chromatography together with tandem-mass spectrometry in patients treated with DOACs.3,7,10 This is the reference quantitative measurement method.
Recently, a new reference method was validated for the measurement of DOAC levels in plasma. It is referred to as ultra-performance liquid chromatography (UPLC) and like its predecessor mentioned above, it is used in tandem with mass spectrometry (UPLC-MS/MS). UPLC is essentially the same methodology as high-performance liquid chromatography (HPLC). The difference is that UPLC can utilize smaller particle sizes (down to 2 microns) at higher column pressures than HPLC. This allows for faster analysis times and higher sensitivity. Liquid chromatography is widely recognized as a superior method for determining drug level concentrations. This technique is much more selective than clot-based assays because these assays are susceptible to interference with other anticoagulants and preanalytic and endogenous conditions that might affect coagulation factors. However, UPLC-MS/MS results are not influenced by the presence of other compounds due to its specificity. As other studies have shown,3,7,10 the chromogenic anti-Xa assays are superior to clot-based assays for DOAC monitoring. This is supported by the finding that chromogenic anti-Xa assay results are more consistent with UPLC-MS/MS results than other coagulation assays. This positive correlation with chromogenic ant-Xa assays is important because most laboratories do not have access to liquid chromatography-mass spectrometry systems for routine testing.6
It should be noted that the calibrators and controls for DOACs have yet to get FDA clearance. Because of this, clinical laboratories may have to rely on the semiquantitative Prothrombin Time (PT) assay for measuring DOAC levels. The sensitivity of the PT test to DOAC concentration varies with the reagent. For example Triniclot PT Excel S was shown to have the highest sensitivity to apixaban at 154 ng/ml. The lowest sensitivity was seen with Siemens Innovin PT reagent at 1364 ng/ml. However, even the most sensitive of these PT reagents fails to provide the needed sensitivity for accurate apixaban measurement.7
An important point to consider when testing DOAC levels in patient plasma is the delay between the last intake of the drug and the sampling of the blood since the results will depend on the time of sampling relative to dose administration. Peak concentration is reached within 1 to 3 hours post dose for apaxiban, 1 to 2 hours for edoxaban and 2 to 4 hours with rivaroxaban.3,7,10
CONCLUSION
The advent of the DOACs such as apixaban, rivaroxaban and edoxaban provide a better long-term alternative to traditional VKA and parenteral anticoagulant therapies for patients suffering from thromboembolic disease or cardiac arrhythmias. The disadvantages of VKAs is their tendency to cross react with many medications and foods, the length of time required to reach a very narrow therapeutic range and their wide range of pharmocodynamic responses at a given blood concentration from patient to patient. For this reason they require constant monitoring and dose adjustments. Parenteral agents are not good long-term solutions due to their expense and their inability to be given orally. Unlike VKAs, DOACs have predictable pharmocodynamic and pharmocokinetic properties and for that reason have little need for monitoring. This is due to their specific mechanism of action. They are inhibitors of factor Xa only whereas VKAs have numerous targets in the coagulation cascade. However, when it is necessary to determine DOAC concentrations, the chromogenic anti-Xa assay is the best linear quantitative assay due to its sensitivity and accessibility to most clinical laboratories. Unfortunately at this time calibrators and controls for apixaban, rivaroxaban and edoxaban have yet to be approved by the FDA, so plasma levels may have to be performed using the semiquantitive Prothrombin Time assay which lacks the sensitivity of the chromogenic anti-Xa. No matter what method a clinical laboratory employs for DOAC measurement, the results should be periodically verified using liquid chromatography in tandem with mass spectrometry which is the reference method for measuring DOAC levels in plasma. As of now relatively few providers are using these new DOACs compared to the more traditional anticoagulant therapies. This is likely due to their unfamiliarity with this new method but as their confidence in DOACs grows hospital formularies will be challenged to appropriately integrate these new drugs as their demand increases.
ACKNOWLEDGEMENTS
Thanks to George Fritsma for his keen editorial eye and encouragement.
- © Copyright 2016 American Society for Clinical Laboratory Science Inc. All rights reserved.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.