


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Larry J. Smith, PhD, SH(ASCP)⇑
- Address for Correspondence: Larry J Smith, PhD, SH(ASCP), Abbott Diagnostics Division – Hematology Business Unit, 4551 Great America Pkwy, Santa Clara, CA 95054
Abstract
Von Willebrand disease (VWD) is considered the most common congenital bleeding abnormality in the world and an accurate diagnosis is often very challenging. Clinical laboratories usually provide screening assays along with more complex assays to aid clinicians with the diagnosis. These assays measure different properties and activities of von Willebrand factor. They may be adversely affected by numerous preanalytical variables and often have variable performance characteristics that may contribute to an inadequate interpretation. This review describes methods used in the laboratory for diagnosing VWD and basic mechanisms of each of these assays. A thorough understanding of the assays and associated preanalytical variables in VWD testing is essential for accurate diagnosis.
ABBREVIATIONS: ADAMTS13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif, DDAVP - 1-desamino-8-D-arginine vasopressin, EC - endothelial cell, VWD - von Willebrand disease, VWF - von Willebrand factor, VWF:Ag - von Willebrand factor antigen, VWF:CBA - von Willebrand factor collagen binding assay, VWF:RCo - von Willebrand factor ristocetin cofactor activity
- INDEX TERMS
- von Willebrand disease
- von Willebrand factor
- ristocetin cofactor
- von Willebrand factor activity
- von Willebrand factor antigen
- collagen binding assay
- von Willebrand factor multimers
INTRODUCTION
Accurate diagnosis of von Willebrand disease (VWD) can often be very challenging for clinicians. In addition to patient and family history, clinicians utilize a panel of assays provided by the clinical laboratory to aid in diagnosis. These most often include an assessment of von Willebrand factor (VWF) activity and antigen levels, along with coagulation FVIII activity levels. The combination of these three assays is necessary for diagnosis of VWD and when used in conjunction with VWF multimer analysis, aids in differentiating subtypes of VWD. The most critical of these assays to the diagnosis of VWD is probably the assessment of VWF activity.
History
Von Willebrand disease is defined as an autosomal inherited bleeding disorder that is caused by either a deficiency of, or a dysfunctional von Willebrand factor (VWF) molecule.1 It is the most common congenital bleeding disorder and occurs at a frequency of 0.1% to 1% in the general population.2 Actual estimates may be higher in the general population since many people with a VWF mutation are asymptomatic or undiagnosed. The disease was first described in 1926 by Dr. Erik von Willebrand in a 5-year-old female who lived in Föglö on the Åland Islands.3 The patient presented with mucocutaneous bleeding, a prolonged bleeding time, and a family history of bleeding. She died in her early teens as a result of increased menstrual bleeding. Four other family members also died due to uncontrolled bleeding diatheses.4 Dr. von Willebrand recognized that this bleeding pattern differed from classical hemophilia and appeared to have an autosomal inheritance pattern. He called this bleeding disorder hereditary pseudohemophilia.3
Synthesis
VWF is a large adhesive protein that is synthesized in endothelial cells and megakaryocytes. In endothelial cells (EC), it is stored in the Weibel-Palade bodies (WPB) and in megakaryocytes in the alpha granules.5 It is composed of multimeric units that range in size to >20 million Daltons.6 VWF is initially synthesized as a large pre-pro VWF monomer composed of a 22-amino acid signal peptide, a 741-amino acid propeptide, and a 2,050-amino acid mature VWF protein.7 When the protein reaches the endoplasmic reticulum (ER), the signal peptide is removed and the protein undergoes dimerization. The dimers are covalently linked together at their carboxyl terminal ends.8 As the protein passes to the Golgi apparatus glycosylation occurs, and the dimers undergo multimerization, increasing in size. The propeptide portion is cleaved by furin (paired basic amino acid cleaving enzyme, aka PACE) resulting in a mature VWF molecule.8 Following cleavage, the mature VWF subunit is tightly coiled and packaged in the WPB. VWF is released from ECs constitutively into the circulating plasma and the subendothelial matrix.2 The unusually large multimers of VWF, which have an increased binding affinity to platelets, are cleaved into smaller subunits by a protease, ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 repeats, member 13), located on the EC surface.9
Structure
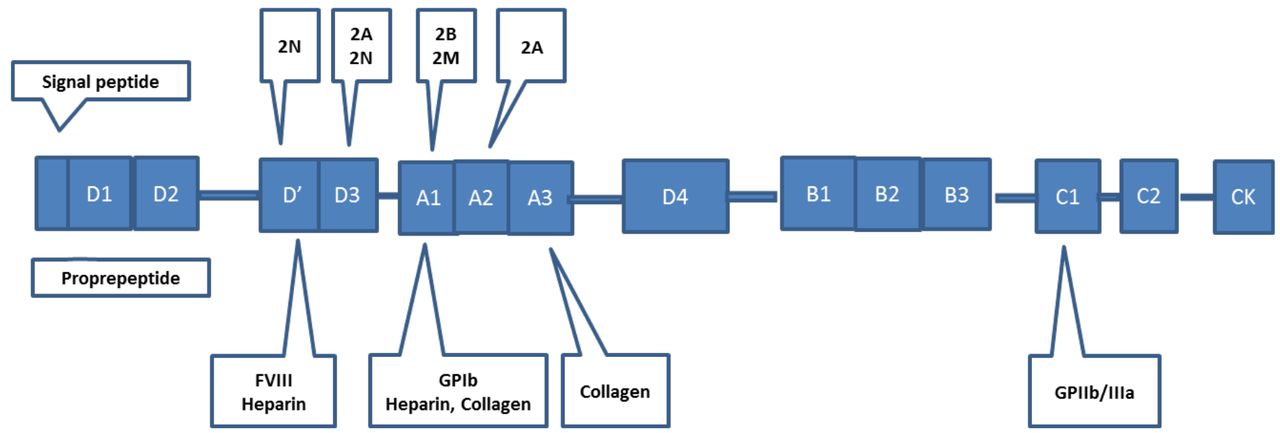
The gene responsible for VWF is located on the short arm of chromosome 12 (12p13.3). It is a large gene that contains 178 kb of genomic DNA consisting of 52 exons.1,10 The coding sequence was identified in 1986. The cDNA is divided into the following domains: D1-D-2-D’-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2 (Figure 1). D1-D2 represents the propeptide region, while the remaining domains represent the mature subunit.2 The clinical heterogeneity seen in VWD reflects the variety of mutations that occur throughout the various domains. In addition, a pseudogene for VWF is located on chromosome 22 and has ~98% homology to the gene located on chromosome 12.1 The pseudogene replicates exons 23-34 with about 3.1% variation in DNA sequence. The presence of the pseudogene may complicate analysis based on polymerase chain reactions (PCRs), which can be avoided by careful design of the gene-specific PCR primers used in the reaction.1
Function
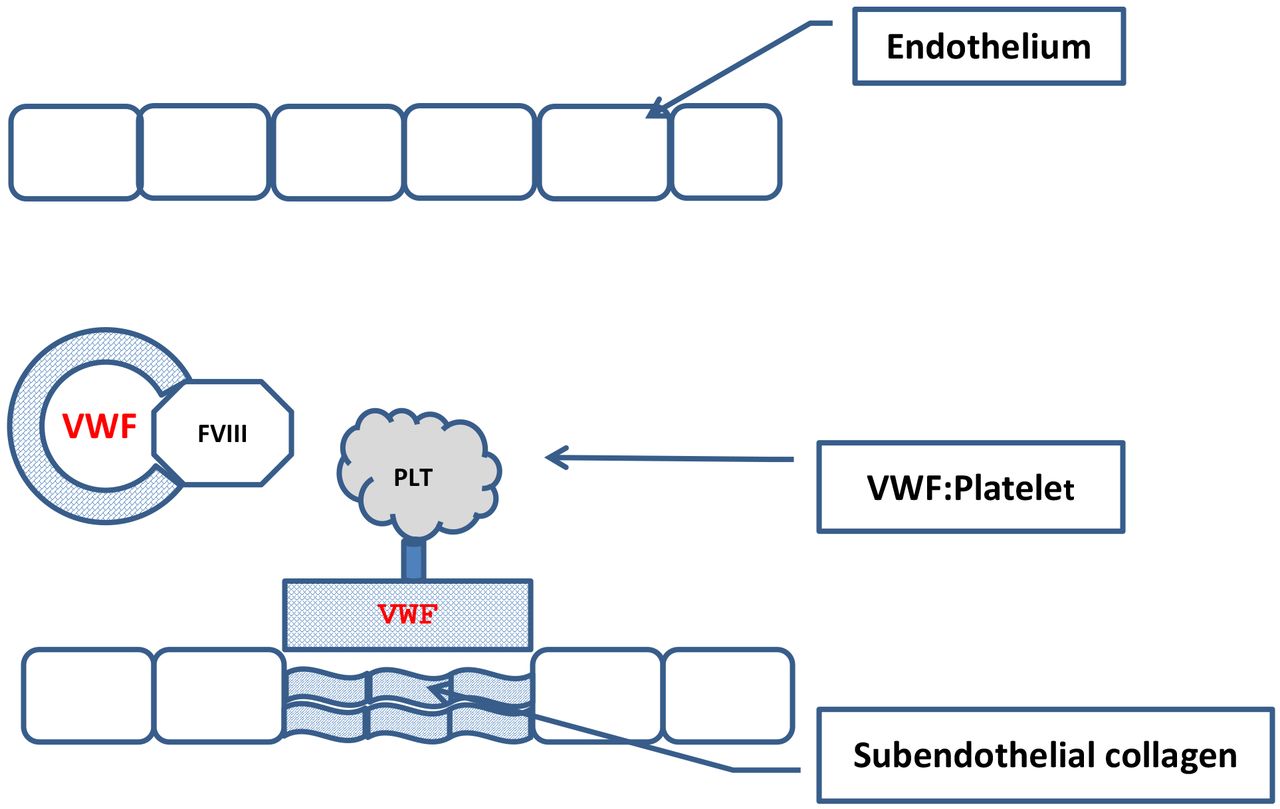
VWF serves several important biologic functions (Figure 2). VWF binds subendothelial collagen microfibrils that are exposed during vascular injury. The bound VWF in turn binds the glycoprotein (GP) Ib portion of the platelet membrane GP Ib/V/IX receptor complex, allowing platelets to adhere to the site of injury. The VWF-platelet combination is essential in areas of high shear stress, capillaries and arterioles, where direct platelet-collagen binding is ineffective. The VWF-platelet combination is termed a “platelet plug.”11 VWF also supports platelet aggregation as it, along with fibrinogen, binds platelet membrane GP IIb/IIIa. Finally VWF is the transport protein for FVIII circulating in the plasma, protecting it from proteolysis and prolonging its half-life in the circulation. This serves to co-localize FVIII to the site of vascular injury allowing it to interact with the proteins in the coagulation cascade.6

The von Willebrand Factor protein showing various functional domains that have been mapped to regions of the cDNA. These show sites of interaction with FVIII, GPIb, GPIIb/IIIa, collagen, and heparin. Various genetic mutations have been mapped to specific regions of the cDNA as shown above, and mutations within these domains result in different clinical expressions of VWD. A mutation in the D’ domain results in type 2N VWD where there is a loss of the FVIII binding ability of VWF. A mutation in the D3 domain results in types 2A and 2N VWD where there is a loss in the platelet binding ability of VWF or the ability to bind to FVIII. A mutation in the A1 domain results in types 2B and 2M VWD where there is either a gain-of-function mutation in the VWF molecule or a loss of the platelet binding ability of VWF. A mutation in the A2 domain results in type 2A VWD where there is a loss of the platelet binding ability of VWF.

Function of VWF. VWF supports platelet adhesion to the damaged endothelial layer and forms a complex with FVIII in the circulation.
Classification of VWD
VWD is an extremely heterogeneous complex disorder that consists of three major categories: type 1 (partial quantitative deficiency), type 2 (qualitative defect), and type 3 (total deficiency). Type 2 is further broken down into four subtypes: subtypes 2A, 2B, 2M, and 2N, (Table 1).
Classification of von Willebrand Disease
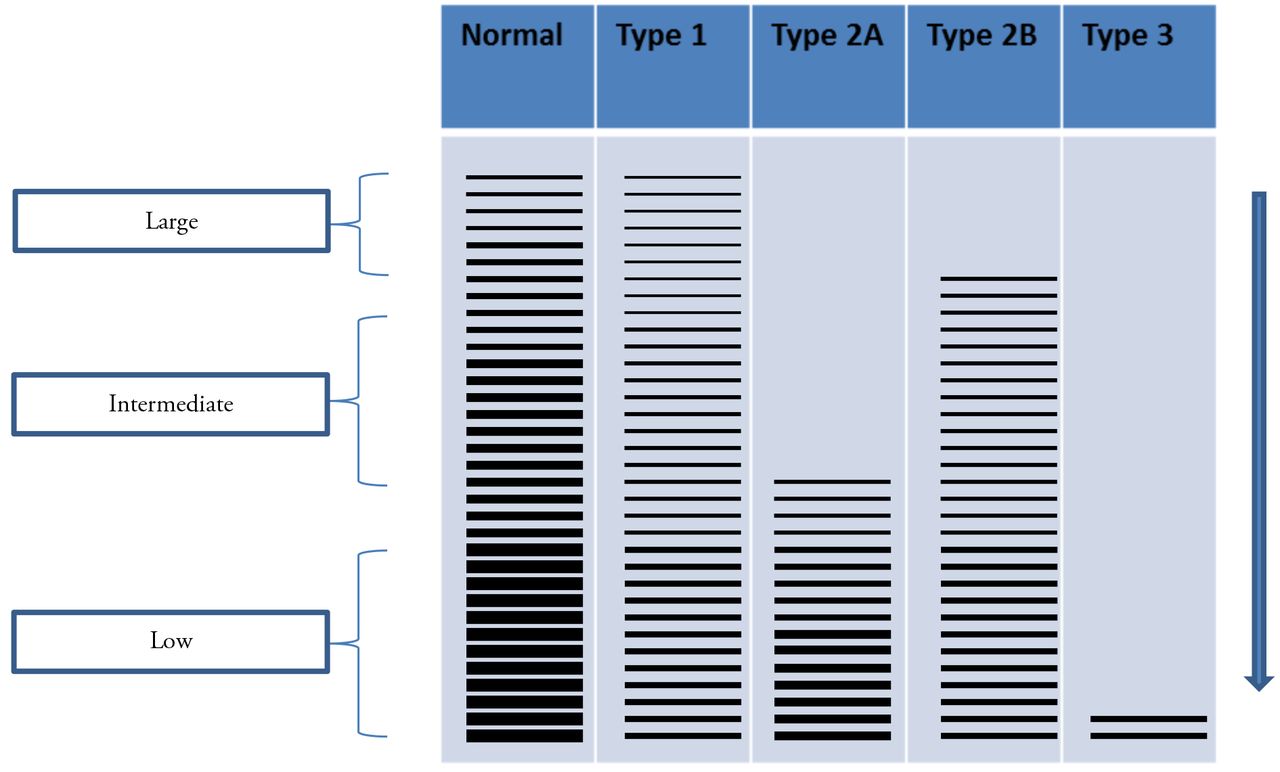
Type 1 VWD is the most common variant of VWD and makes up about 70-80% of the cases. It is characterized by a mild to moderate decrease in VWF as measured by the VWF antigen and activity levels. The VWF antigen:VWF activity ratio is >0.7. In addition there is a corresponding decrease in circulating FVIII levels. On SDS-agarose electrophoresis of VWF multimers, high, intermediate, and low molecular weight multimers are all present; however there is a proportional decrease in the amount of protein present (Figure 3). It is further characterized by a heterozygous mutation that leads to the partial quantitative deficiency of VWF. The quantitative deficiency may be due to increased retention of VWF in ECs, increased clearance from plasma, or decreased production of VWF.12
Patients presenting with low levels of VWF (30-50 IU/dL) are common and may pose a significant challenge for a laboratory diagnosis of type 1 VWD. Even though the VWF levels are low and these patients often present with bleeding symptoms, they do not meet the criteria for a diagnosis of VWD type 1 (<30 IU/dL). This group of patients should be considered to have “low VWF” rather than a diagnosis of type 1 VWD.13

VWF multimers. Low-resolution gel (0.65% agarose) demonstrating VWF multimer distribution. Lane 1 shows normal plasma as a control. Lane 2 shows type 1 VWD with all multimers sizes present, but reduced concentration. Lane 3 shows type 2A VWD with a loss of the high and intermediate multimers. Lane 4 shows type 2B with a loss of only the large multimers. Lane 3 show type 3 VWD with no detectable multimers
Type 3 VWD is a rare form of VWD, occurring in less than 1% of patients. This form is characterized by levels of VWF antigen and activity less than 10 IU/dL.14 This is due to a homozygous or doubly heterozygous mode of inheritance. The circulating FVIII levels range from 2-10 IU/dL. In addition, the multimers cannot be visualized (Figure 3).
Type 2 VWD is divided into 4 subtypes and is due to a qualitative defect in the VWF protein. These include subtypes 2A, 2B, 2M, and 2N.
Subtype 2A VWD accounts for approximately 10-15% of the cases of VWD and is due to an autosomal dominant mode of inheritance. Mutations leading to the defective VWF protein in subtype 2A appear to cluster in the A2 domain near the cleavage site for ADAMTS13 and are also seen in other domains (Figure 1).12 The defect results in increased proteolysis of the protein or an abnormal assembly and secretion of the large multimers.1 Patients with subtype 2A VWD present with moderate to severe mucocutaneous bleeding due to decreased platelet adhesion. The levels of VWF antigen and FVIII are normal to decreased and the VWF activity level is disproportionately low compared to the antigen. The VWF:RCo/VWF:AG ratio is <0.7. Multimer analysis reveals loss of both the high and intermediate molecular weight multimers (Figure 3).
Subtype 2B VWD accounts for about 5% of the cases of VWD and also results from an autosomal dominant mode of inheritance. The mutation results in increased binding of the mutated VWF to the platelet GPIb receptor. This is referred to as a “gain-of-function” mutation and leads to spontaneous binding of VWF to platelets in the circulation, thrombocytopenia, and loss of only the high molecular weight multimers (Figure 3). Thrombocytopenia may not always be present.14 The VWF antigen level is decreased or normal and the VWF activity is disproportionately decreased compared to the VWF antigen level. The VWF:RCo/VWF:AG ratio is <0.7. Interestingly, patients presenting with subtype 2B VWD demonstrate a rather vigorous agglutination response to low doses of ristocetin compared to normal individuals. Therefore, when doing platelet aggregation studies it is recommended to use high, intermediate, and low doses of ristocetin (RIPA, ristocetin-induced platelet aggregation) to identify this subtype of VWD.
A “gain-of-function” mutation may also occur in the GPIb receptor of platelets rather than in the VWF molecule.15 This defect is called platelet-type or pseudo-VWD and patients present with identical laboratory findings seen in subtype 2B VWD. It is important to differentiate between patients presenting with subtype 2B VWD and platelet-type or pseudo-VWD as the treatment options would be different for these individuals.
Subtype 2M VWD is seen in <5% of the cases of VWD and is characterized by a decreased ability of VWF to bind to the GPIb receptor of platelets. The mutation occurs in the A1 domain. VWF activity levels are disproportionately lower than the VWF antigen levels with a ratio usually <0.7.14 In subtype 2M there is no loss of the high or intermediate molecular weight multimers, however they are not able to bind to platelets, which leads to mucocutaneous bleeding in these patients. The levels of FVIII are normal to decreased depending on the level of circulating VWF antigen.
Subtype 2N VWD is relatively rare. The mutation in subtype 2N VWD results in an inability of VWF to bind and protect FVIII in the circulation.16 As a result, the half-life of FVIII in the circulation decreases from 8-12 hours to about 2 hours. The ability of VWF to adhere to platelets is not affected, so patients with subtype 2N VWD do not present with mucocutaneous bleeding, but rather bleeding into the joints and soft tissues. These patients can easily be misdiagnosed as having hemophilia due to the low levels of circulating FVIII.
Laboratory Diagnosis
Due to the complex heterogeneity of VWD, the ability to make an accurate diagnosis is important. Clinical bleeding ranges from asymptomatic to symptomatic and will vary depending on the type of VWD. In type 1 VWD most cases will require a hemostatic challenge such as trauma or surgery to trigger the bleeding event. In types 2 and 3 VWD, bleeding may be more severe and may occur spontaneously in some cases.
At minimum, three tests are included in the current standard practice for laboratory diagnosis of VWD. These include (1) von Willebrand factor antigen level (VWF:Ag), (2) von Willebrand factor activity (VWF:Act or VWF:RCo) level, and (3) circulating level of FVIII (FVIII:C) (Table 2 and 3).
Laboratory assays for von Willebrand Disease Diagnosis
Classification of VWD by screening assays
VWF Antigen Assay
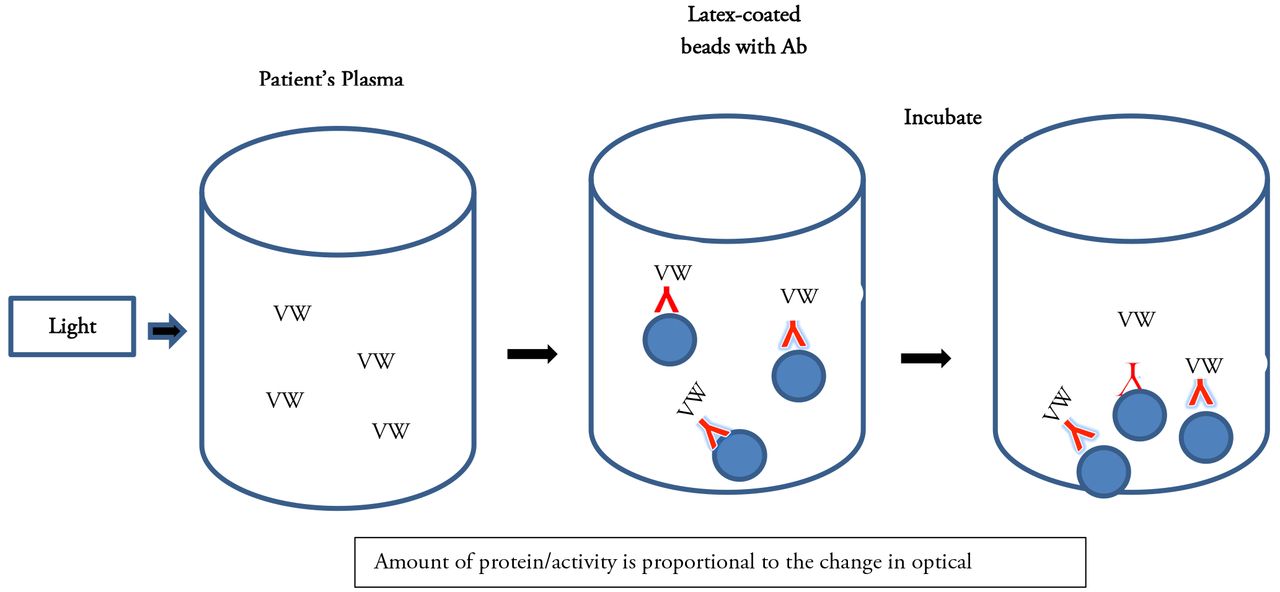
The VWF:Ag is most often an immuno-based assay that can be performed as an ELISA (enzyme-linked immunosorbent assay) method or as an automated turbidimetric method using latex beads coated with antibody to VWF (Figure 4). If the VWF antigen is present in patient plasma, it will bind to the VWF antibody attached to the bottom of a microtiter plate in the ELISA assay or to the antibody-coated latex beads in the turbidimetric method. VWF is then quantified based on the amount of antigen/antibody binding that occurs. The VWF:Ag assay measures the amount of protein that is present but does not assess the functional status of the protein. The normal range is 50-150 IU/dL but may vary with individual laboratories.14,17

Latex immunoturbidometric assay. Latex beads are coated with a monoclonal antibody that binds to VWF.

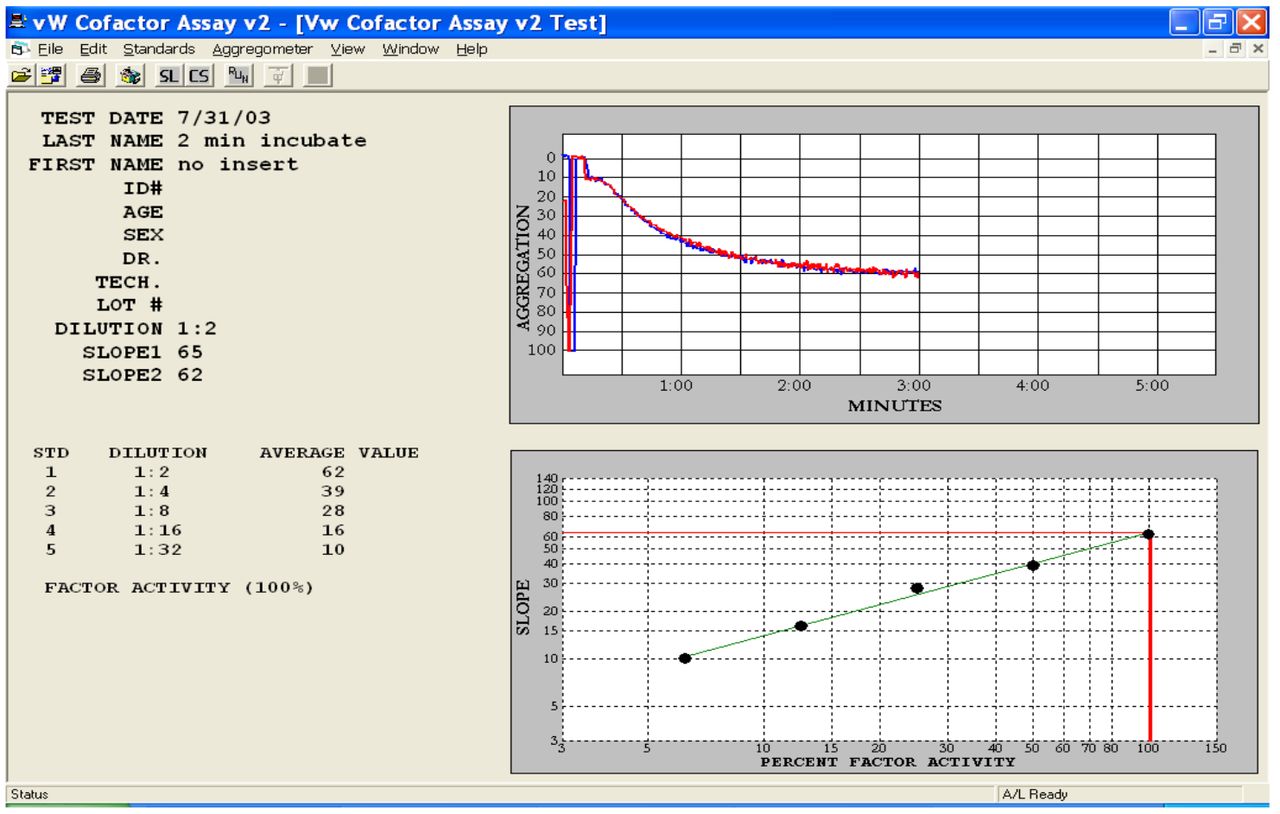
Ristocetin cofactor assay measures the ability of VWF in plasma to agglutinate platelets in the presence of Ristocetin. The slope of the aggregation pattern is measured and the activity of VWF is determined from a calibration curve. (From Chrono-log VW Cofactor assay software, Chrono-Log Corporation, Havertown, PA).
VWF Activity Assay
Von Willebrand activity can be measured by a number of different methods including; 1) the ristocetin cofactor assay (VWF:RCo), the gold standard or reference method for measuring VWF activity; 2) automated immunoturbidimetric assays 3) a chemiluminescence assay; or 4) an enzyme-linked immunosorbent assays (ELISA); or 5) flow cytometry.
The VWF:RCo assay estimates the functional properties of VWF in a patient's plasma. Ristocetin, an antibiotic, induces binding of VWF to the GPIb receptor to platelets. This leads to VWF-dependent agglutination of platelets. Diluted patient plasma containing VWF is added to normal platelets (formalin-fixed platelets or normal donor platelets) in the presence of ristocetin. Platelet agglutination is measured using a platelet aggregometer. The slope of the aggregation tracing is determined and compared to a standard calibration curve to determine the VWF activity (Figure 5). The VWF:RCo is considered a “functional assay”, because it measures the interaction of VWF with the GPIb platelet receptor.17 In vivo, this interaction results from high shear rather than the presence of ristocetin. Due to high intra- and inter-assay variation (coefficient of variation 10-30%), many laboratories have looked for alternative methods to the VWF:RCo assay as a way of measuring VWF activity. Additional limitations of the VWF:RCo include a decreased sensitivity to the loss of high molecular weight VWF multimers.18 Several immunoturbidimetric assays have been developed, which measure VWF activity that do not require ristocetin. Even though these assays measure VWF activity, they are not “true” functional assays. They measure an antigen;antibody reaction rather than the actual binding of VWF factor to platelets. All are automated and have improved precision compared to the VWF:RCo assay.
One immunoturbidimetric assay uses latex-coated beads with a specific monoclonal antibody that binds to the domain in the VWF molecule where the platelet GPIb receptor binds and does not require ristocetin (Instrumentation Laboratories, Bedford, MA, USA) (Figure 4). The VWF activity is determined based on changes in optical density that occur due to the degree of bead agglutination and is directly proportional to the level of VWF activity in the patient's plasma.19 This assay has been shown to have a much better intra- and inter-assay variation (<10%) compared to the VWF:RCo assay. In addition, it was shown to have an improved lower detection limit (3 IU/dL) compared to the VWF:RCo assay.19 The VWF-RCo assay or the immunoturbidimetric assay, when used in conjunction with supplemental VWF assays, it will improve diagnostic accuracy for diagnosis and monitoring treatment in VWD.20
Another immunoturbidimetric assay uses a recombinant form of the VWF platelet membrane receptor (GPIbα) containing two gain-of-function mutations and an antibody against GPIbα captured onto polystyrene particles (Siemens Healthcare Diagnostics, Marburg, Germany). VWF in the sample binds to the polystyrene particles leading to a change in turbidity. The change in turbidity correlates to VWF activity.18, 21 The gain-of-function mutations allow for binding of VWF in the absence of ristocetin.22 This assay has also has been shown to have better intra- and inter-assay variation (<10%) compared with the VWF:RCo assay.18 This method was also shown to be sensitive to both quantitative and qualitative defects of VWF and to have acceptable performance characteristics for diagnosing and monitoring the treatment of VWD.
An automated chemiluminescent immunoassay (Instrumentation Laboratories, Bedford, MA, USA) can also be used to measure VWF activity by a two-step process. In the first step, the sample is mixed with a ristocetin containing assay buffer and magnetic particles coated with a recombinant fragment of GP1bα. A specific monoclonal antibody orients the GP1bα fragment to interact with VWF in the presence of ristocetin. VWF binds to the magnetic particles and is proportional to the activity of VWF. In the second step, an anti-VWF monoclonal antibody labeled with isoluminol is added and chemiluminescence is measured and reported as relative light units (RLUs) corresponding to the activity of VWF.23 This method demonstrated good precision (<8%) and lower limit of detection (0.2 IU/dL).24
VWF:Act can also be measured by ELISA assays and flow cytometry. ELISA assays utilize a purified murine anti-vWF monoclonal antibody that recognizes a functional epitope of VWF.25 VWF activity is estimated by from a standard curve. Flow cytometric analysis has been studied and shown to be useful in diagnosing and managing VWD. In a study performed by Giannini et al, ristocetin-induced binding of VWF to platelets was measured and shown to be sensitive, simple and rapid for diagnosis and monitoring in VWD.15 Although a small study, it showed good correlation with existing methods.
The collagen binding assay (VWF:CBA) is an ELISA-based assay that measures the ability of VWF to bind to collagen and is associated with the high molecular weight multimers (HMWM) of VWF. The HMWMs of VWF are important in platelet adhesion. When used in conjunction with the VWF:Ag assay, the VWF:CBA has been shown to aid in the differentiation of VWD Type 1, subtype 2A and subtype 2B. The VWF:CBA is not a replacement for the VWF:RCo assay since the two assays measure different functions of VWF. However, the VWF:CBA has been shown to be more sensitive than the VWF:RCo to the loss of the HMWs and has a lower CV. In addition, some patients with a defect in the collagen binding function of their VWF may have a normal VWF:RCo and this defect would only be picked up by using the VWF:CBA assay.
Factor VIII Assay
FVIII activity assay (FVIII:C) is a clot-based assay that measures the ability of various dilutions of patient plasma to correct the activated partial thromboplastin time of a commercially prepared factor VIII-deficient plasma compared to normal plasma.26 Since VWF protects FVIII in the circulation, measuring levels of circulating FVIII is generally included in the workup of VWD. The ratio of FVIII:C to VWF:Ag is approximately 1.0 for all subtypes of VWD except for subtype 2N.
VWF Multimer Assay
VWF multimer analysis assesses the size distribution of VWF multimers and aids in differentiating subtypes of VWD. It consists of electrophoresis to separate the VWF multimers on an agarose gel. Separation is followed by fixation and visualization of the protein on the gel or transfer to a membrane (blotting). The first step involves the use of non-reducing agarose gel electrophoresis where VWF multimers are separated into a series of bands based on their molecular weight. Laboratories can run high-, medium- or low resolution gels. Low resolution gels (<1.0% agarose) are superior for separating the largest VWF multimers, whereas, higher resolution gels (>1.0%) give better resolution of VWF triplet structure.27 Following electrophoresis, the VWF multimers on the gel are incubated with an antibody specific for VWF and visualized by autoradiography (Figure 3). The multimers may also be transferred (blotted) onto a membrane such as nitrocellulose, nylon or polyvinylidene difluoride and visualized immunologically by colorimetry, chemiluminescence or fluorescence. In addition to visualization, quantification of the bands by densitometry can be performed. Multimer analysis is crucial in diagnosing subtypes 2A and 2B VWD where there is a loss of the high and/or high and intermediate molecular weight multimers and is usually not performed in type 1 VWD where the VWF:RCo/VWF:Ag ratio is >0.7. VWF multimer analysis is usually performed by specialized labs requiring interpretation by skilled personnel. In addition, these assays are not standardized.
Additional assays can be used in VWD testing such as the VWD platelet-binding assay to differentiate between subtype 2B VWD and platelet-type VWD; and VWD FVIII binding assay to diagnose subtype 2N VWD. These assays would be performed in highly specialized laboratories.
Clinical presentation in VWD
Bleeding symptoms in VWD range from asymptomatic to severe and are characterized by easy bruising, epistaxis, gingival bleeding, menorrhagia, prolonged bleeding from wounds, post-surgical and post-dental procedures, and gastrointestinal bleeding. In type 1 VWD, bleeding symptoms range from asymptomatic to mild and depend on the level of VWF present. In type 3 VWD, patients present with severe mucocutaneous bleeding and bleeding into the joints and soft tissue. This bleeding pattern might easily be confused with hemophilia due to the low levels of FVIII and hemarthrosis. Bleeding symptoms in type 2 VWD vary between the different subtypes and range from mucocutaneous bleeding seen in 2A, 2B and 2M to patterns suggestive of mild-to-moderate hemophilia seen in 2N.28
Preanalytical Variables
A number of preanalytical variables should be considered when testing and interpreting VWF assays by both the clinician and the laboratory staff. These include collection technique, specimen preparation, storage and transport. Cold storage of citrated whole blood prior to centrifugation has been shown to lead to falsely low levels of VWF.29 Since VWF is an acute phase reactant, stress, illness, strenuous activities, medications, hyperthyroidism, oral contraceptive therapy, hormone replacement therapy, and pregnancy may mask an actual deficiency of VWF.30 Age and race are also associated with elevated levels of VWF.31 In most laboratories, a general reference interval is used for VWF testing that includes a mixture of different ABO blood groups. Decreased levels of VWF are associated with ABO blood group O individuals (~25% lower than non-ABO individuals, 41-179 IU/dL).32 Non ABO blood groups A, B, and AB range from 55-267 IU/dL, 65-275 IU/dL, and 73-271 IU/dL, respectively.1,32
Treatment options in VWD
Treatment options in VWD depend on the type of VWD that the patient has and consideration of other comorbidities. For congenital deficiencies, the goal is to restore the level of VWF (and FVIII) to hemostatic levels. This can be achieved through the use of Desmopressin (1-desamino-8-D-arginine vasopressin, or DDAVP) in type 1 VWD and some cases of type 2 VWD; and, factor replacement therapies (Humate-P, Alphanate). DDAVP is a synthetic analogue of antidiuretic hormone that stimulates the release of VWF from endothelial cells.12 Humate-P and Alphanate contain both VWF and FVIII. A patient's response to these therapies can be monitored using any of the screening assays mentioned above. Additional therapies used to treat VWD include antifibrinolytic therapy (e-aminocaproic acid and tranexamic acid) topical agents, and estrogen.12
- © Copyright 2017 American Society for Clinical Laboratory Science Inc. All rights reserved.
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.