


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Karissa Wang, CAN⇑
- Address for Correspondence: Karissa Wang, CNA, Nora Eccles Harrison Cardiovascular Research & Training Institute, 95 S 2000 E, Salt Lake City, UT 84112, 801-317-5280, wangkarissa{at}gmail.com
Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) are genetic elements that function with CRISPR-Associated (Cas) proteins as an adaptive immune system to foreign genetic material in prokaryotic organisms. The CRISPR-dCas9 system is modified to suppress gene transcription. Methicillin-Resistant Staphylococcus aureus (MRSA) is a dangerous human pathogen that is resistant to beta-lactam antibiotics. This is due to the mecA methicillin resistance gene coding for penicillin binding protein 2A (PBP 2A), which inhibits the activity of beta-lactam antibiotics. Two CRISPR-dCas9 systems were designed to target the promoter region of mecA in MRSA to suppress transcription of the gene. A cefoxitin disk diffusion test showed that the target on the coding strand significantly reduced antibiotic resistance in MRSA, whereas the target on the noncoding strand did not. An oxacillin microbroth serial dilution was used to confirm disk diffusion results. The CRISPR system targeting the coding strand was the only one to reduce antibiotic resistance and thus was chosen for continued testing. mecA gene expression levels were analyzed using Reverse Transcriptase Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR). Results showed that mecA gene expression in the CRISPR-treated sample was reduced to 0.230 fold of the value in the control, representing a 77% decrease in gene transcription. The 77% decrease in gene expression was not enough to make MRSA clinically susceptible to betalactam antibiotics.
ABBREVIATIONS: BLAST - basic local alignment search tool, bp - basepairs, Cas - CRISPR-associated, cDNA - complementary DNA, CLSI - Clinical Laboratory Standards Institute, Cq - quantification cycle, CRISPRs - clustered regularly spaced short palindromic repeats, E.coli - Escherichia coli, M-MLV RT - Moloney Murine Leukemia Virus Reverse Transcriptase, MIC - minimum inhibitory concentration, MRSA - methicillin-resistant Staphylococcus aureus, NCBI - National Center for Biotechnology Information, PAM - protospacer adjacent motif, PBP 2A - penicillin binding protein 2A, RT-qPCR - reverse transcriptase quantitative real-time polymerase chain reaction, tracr - trans-activating CRISPR, UDG - uracil DNA glycosylase
- INDEX TERMS
- Clustered Regularly Interspaced Short Palindromic Repeats
- Methicillin-Resistant Staphylococcus aureus
- Drug Resistance
- Microbial
- Penicillin-Binding Proteins
INTRODUCTION
Methicillin-Resistant Staphylococcus aureus (MRSA) are responsible for many human maladies including necrotizing pneumonia and nosocomial infections.1 Over 70,000 invasive MRSA infections occurred in 2014, with more than 9,000 of these resulting in death.2 MRSA infections are particularly difficult to treat due to the bacteria's resistance to beta-lactam antibiotics.3 Betalactam antibiotics such as penicillin, methicillin, oxacillin, and cefoxitin function by prohibiting cell wall formation in bacteria. MRSA possesses the mecA methicillin resistance gene which produces Penicillin Binding Protein 2A (PBP 2A), a mutated protein that prevents beta-lactam antibiotics from functioning correctly.
Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPRs) are DNA segments consisting of spacer sequences separated by repeating basepair sequences.4 CRISPRs, along with CRISPR-Associated (Cas) proteins, are used by prokaryotic organisms as adaptive immune systems to target foreign genetic material. This process begins when foreign DNA from invading viruses or plasmids is integrated onto the CRISPR gene locus as spacer sequences separated by repeats.5 When the CRISPR locus is transcribed, a CRISPR endonuclease complex complementary to the invader DNA sequence is created. In a common type of CRISPR system, this complex, called CRISPR-Cas9, binds to and cleaves the invader gene sequence, thus preventing viral replication. CRISPRs have been adapted for use in genetic engineering to target specific sequences determined by researchers in order to inactivate or suppress genes. Since the discovery of CRISPRs, researchers have used the technology to edit human embryos, initiate gene drives in mosquitos, and kill specific bacterial cells by causing irreparable DNA damage.6-8 CRISPR-dCas9 is a system modified to be catalytically inactive, resulting in the complex binding to the gene, but not cleaving it.9 Both CRISPR-Cas9 and CRISPR-dCas9 systems prevent transcription of a target gene, but the latter leaves the genes intact. This study employed two CRISPR-dCas9 systems in the form of plasmids, each designed to target a different site on the mecA methicillin resistance gene. CRISPR-dCas9 systems were used instead of CRISPR-Cas9 systems in hope of suppressing antibiotic resistance without killing the target bacteria. The phenotypic effects of the systems were examined using Minimum Inhibitory Concentration (MIC) testing and genotypic effects were examined using Reverse Transcriptase Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR). Treatment by one of these CRISPR-dCas9 systems significantly reduced mecA gene expression in MRSA by 77%, yet this suppression was not enough to confer susceptibility to beta-lactam antibiotics.
MATERIALS AND METHODS
Plasmid Design
The mecA gene sequence (accession number KF058908.1) for MRSA ATCC 43300 was downloaded from the National Center of Biotechnology Information (NCBI), then scanned for potential CRISPR target sites. Potential sites were ranked based on proximity to the gene promoter and location on coding or complement DNA strand. Each target was a 30 basepair (bp) region upstream of a Protospacer Adjacent Motif (PAM), a 3 bp NGG sequence essential to target recognition. Previous research has shown that PAMs near the Pribnow Box, a region 10 to thirty-five bp upstream of the promoter, function best. The same study also suggested that target sites on the coding strand of DNA are more efficient than targets on the noncoding strand.9 Two sites were chosen: site 43 (S43) on the coding strand of mecA and site 46 (S46) on the noncoding strand. A Basic Local Alignment Search Tool (BLAST) test was run to ensure the target sites would be specific to the mecA gene. Three groups of CRISPR-dCas9 plasmids were produced: one with the S43 target, one with the S46 target, and a control with no target site (mock). Each plasmid contained CRISPR-associated genes, Trans-Activating CRISPR (tracr) genes, a CRISPR target sequence flanked by repeats, and a chloramphenicol resistance gene for selection purposes, adding up to 9326 bp per plasmid.
Bacterial Transformation
Plasmids were cloned in Escherichia coli (E. coli) SA30B. This E. coli strain is modified so that it does not add methyl tags to DNA, which is critical to plasmid incorporation into S. aureus. E.coli cells were made electrocompetent by chilling cells on ice for 15 minutes, spinning at 3700 rpm for 20 minutes, suspending in ultrapure H2O, spinning for 20 minutes at 3700 rpm, resuspending in cold 10% glycerol, spinning for 20 minutes at 3700 rpm, and resuspending in cold 10% glycerol. Plasmids were electroporated into competent E.coli cells at 25 μF, 2.5 kV of electricity, and 200 Ω. Cells were then plated on chloramphenicol agar and incubated overnight. Plasmids were extracted from E. coli SA30B using a Qiagen Plasmid Mini Kit. Electrocompetent MRSA cells were made according to previously published instructions.10 1-2 μg of DNA was electroporated into MRSA at 25 μF, 2.3 kV of electricity, and 100 Ω. 1 mm electroporation cuvettes were used. Three collections of MRSA were electroporated: MRSA with the S43 plasmid, MRSA with the S46 plasmid, and MRSA with the mock plasmid. These groups of MRSA were then plated on chloramphenicol agar to select for only the bacteria that had accepted the CRISPR-dCas9 plasmids. The projected transformation efficiency was 1*10^5 CFU/5 μg DNA.11
MIC Testing
Successfully transformed MRSA were used in two antibiotic tests to determine the Minimum Inhibitory Concentrations (MICs). A disk diffusion test was performed in quadruplicate on Kirby-Bauer plates with 30 μg cefoxitin disks. Results of the disk diffusion test were analyzed by measuring the zone of inhibition. Transformed MRSA were also tested using oxacillin microbroth serial dilutions in duplicate. The bacteria were used to inoculate tubes containing 16 μg/mL, 8 μg/mL, 4 μg/mL, 2 μg/mL, 1 μg/mL, and 0.5 μg/mL of oxacillin. The disk diffusion and microbroth serial dilution tests were carried out in compliance with standard procedures outlined by the Clinical & Laboratory Standards Institute (CLSI) in M02-A1212 and M07-A10,13 respectively. The results of these tests were interpreted according to the breakpoints listed in the CLSI procedures to determine antibiotic susceptibility or resistance.
RNA Extraction and Reverse Transcription
Transformed bacteria were cultured overnight, then incubated for 3 hours in the presence of 4 μg/mL of oxacillin to induce mecA gene expression. An Aurum Total RNA Mini Kit was used to extract total RNA from 2 mL 1.5 OD MRSA cultures. Cultures were centrifuged and added to lysozyme and lysis solution, mixed with 70% isopropanol, and then filtered and eluted from RNA binding columns. DNase included in the kit was added to degrade any DNA that might have remained in the sample. The extracted RNA was quantified and assessed for purity using an Epoch 2 Microplate Reader. The average concentration was 29.26 ng/μL and the purity was adequate for each sample.
A complementary DNA (cDNA) library was created from the total MRSA RNA using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT). The appropriate amount of RNA was brought to a volume of 10 μL with distilled water to achieve a concentration of ~12 ng/μL. 1 μL of the appropriate 2 μM reverse primer was added to each sample, along with 1 μL of 10 mM DNTPs. The mixture was then heated to 65°C for 5 minutes and quickly chilled on ice. The contents were centrifuged, then mixed with 4 μL of 5X first-strand buffer (250 mM Tris-HCl, 375 mM KCl, 15 mM MgCl2), 2 μL of 0.1 M DTT, and 1 μL of 40 units/μL RNaseOUT. The mixtures were mixed and incubated at 37°C for 2 minutes. 1 μL (200 units) of M-MLV RT was added before samples were incubated for another 50 minutes at 37°C. The reaction was inactivated by heating at 70°C for 15 minutes. Samples were stored at −20°C. This procedure was carried out for both the mecA gene and the 16s rRNA housekeeping normalizer control gene, which was to act as an internal control of differences in relative expression and loading of DNA.
Expression Analysis
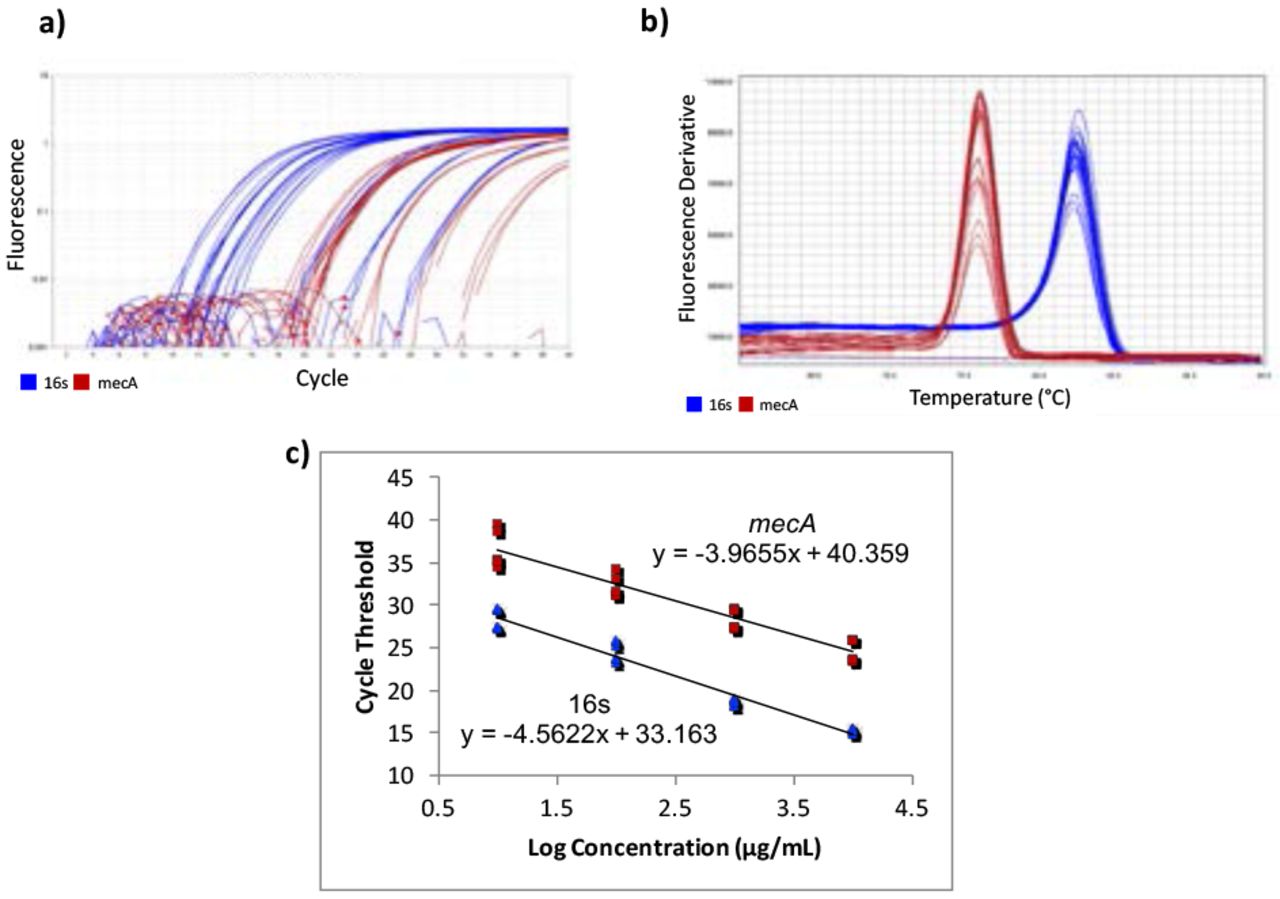
Gene-specific primers for mecA were designed using Benchling14 [F-ATCCACCCTCAAACAGGTGAAT, R-GGAACTTGTTGAGCAGAGGTTC; 139 bp], with an amplicon of 139 bp. Primers from a previous study15 were used for the housekeeping normalizer gene, 16s rRNA [F-CGTGCCTAATACATGCAAGTC, R-CCGTCTTTCACTTTTGAACCA; 174 bp]. Quantitative Real-Time Polymerase Chain Reaction (qPCR) was used to quantify the cDNA. A five-point standard curve was created using serially diluted MRSA cDNA samples. The standard curve was run twice, in triplicate each time. Concentrations in the standard curves ranged from 0.0037185 ng/μL to 37.185 ng/μL (Figure 1c). 10 μL of PowerUp SYBR Green Master Mix (dye, DNA polymerase, dNTPs, passive reference dye, and buffer), 0.6 μL of 10 μM forward primer, 10 μM 0.6 μL of reverse primer, 7.8 μL of H2O, and 1 μL of cDNA were mixed in a standard 96-well 0.2-mL block reaction plate. qPCR was run on a Quantstudio 3 thermocycler programmed to heat wells to 50°C for 2 minutes in order to activate the heat-labile uracil-DNA glycosylase (UDG) and then 95°C for 2 minutes to activate the AmpliTaq Fast DNA polymerase. It then subjected the plate to 40 cycles of 95°C heat for 15 seconds to denature the cDNA, then annealing temperatures of the primers for 15 seconds, and then 72°C for 1 minute to extend the sequences. The annealing temperatures for mecA and 16s primers were 61°C and 60°C, respectively. After amplification, DNA amplicons were heated until they dissociated into single strands. This provided a way to look at the specificity of the reaction to ensure that only the desired product was amplified by melt curve analysis (Figure 1b). Ideally, melt curves contain only one peak to indicate the presence of only one product. Multiple peaks indicate the presence of erroneous DNA products. Two independent experiments were performed, each with two RNA extractions that each tested for 16s and mecA expression levels in triplicate (Table 1). No-Template Controls (NTCs), samples with all reagents present except for cDNA, were also subjected to treatment to show whether contamination was present. The quantification cycle (Cq) of all NTCs was undetermined, meaning there was no contamination in the samples.
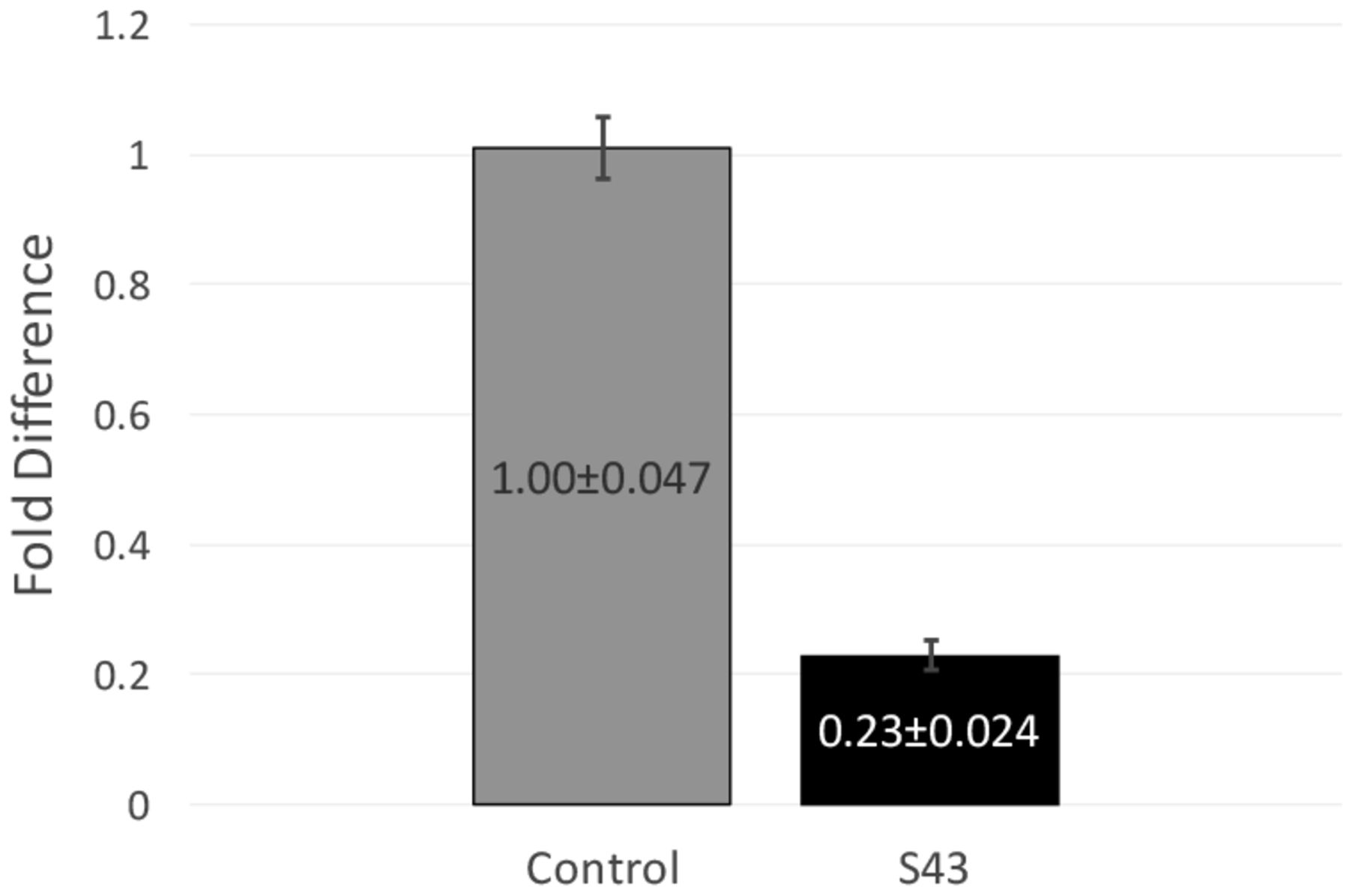
RT-qPCR data and fold difference calculations. Two independent RT-qPCR runs were performed, each with two RNA extractions that each tested for 16s and mecA expression levels in triplicate. Using the comparative quantification standard curve method, the fold difference in bacterial gene expression was given by the ratio of the efficiency of the target gene raised to the ΔCt of the target to the efficiency of the normalizer gene raised to the ΔCt of the normalizer. Efficiencies were calculated from the slopes of the standard curves. Calculations showed an average fold difference of 0.230, meaning that the S43 CRISPR-dCas9 treated MRSA had a significant decrease of 77% in mecA gene expression.
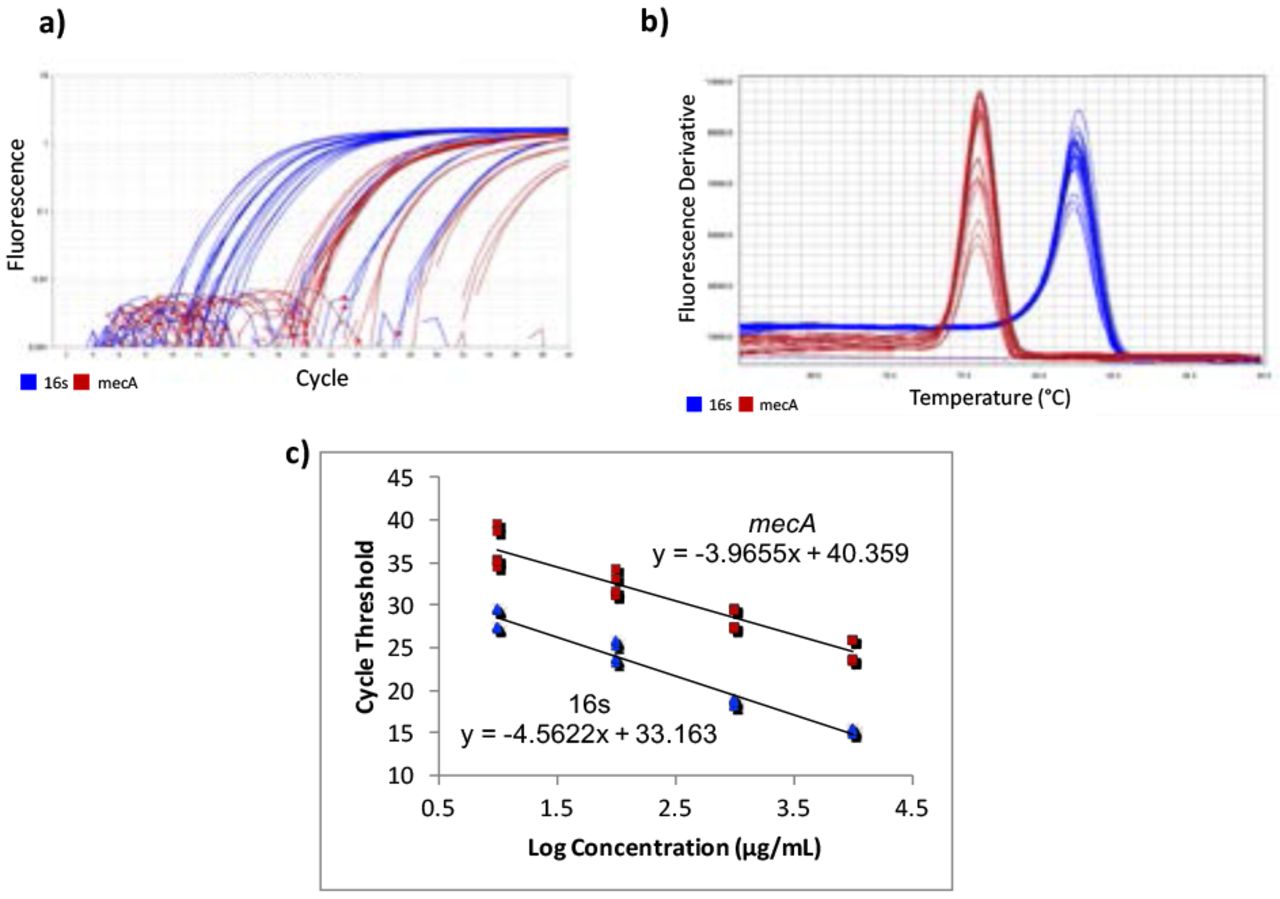
RT-qPCR amplification plots, melt curves, and standard curves. Two independent RT-qPCR experiments were run, with the standard curve in triplicate each time. mecA data is shown in red and 16s data in blue. a) The amplification plot shows that desired precision and amplicon specificity were achieved. b) A smooth, single-peaked melt curve shows satisfactory amplicon specificity for both 16s RNA and mecA. c) The standard curves show log concentrations of MRSA mRNA plotted against quantification cycle values. The slopes are used to describe the efficiency of each target, where an ideal efficiency would yield a slope of −3.3.
Data Analysis
QuantStudio Analysis Software (version 1.3) and Cq values were used to create standard curves. The 1:10,000 dilution for the mecA standard curve contained too low of a DNA concentration, causing it not to amplify consistently. This showed that the lower limit of detection was greater than 0.0037185 ng/μL, so the 1:10,000 dilution was not plotted on the final standard curve.
The comparative quantification standard curve method was used to calculate the fold difference between S43-transformed and mock-transformed bacteria. This method allows the researcher to control sample variability with an endogenous control and also controls for differences in reaction efficiencies of RT and Taq enzymes. This method is outlined in the Real-Time PCR handbook from Life Biosciences.16 The comparative quantification standard curve method employs the equation: fold difference = (Etarget)^(ΔCqtarget) / (Enormalizer)^(ΔCqnormalizer), where E is the efficiency from the standard curve (E=10^(-1/slope)) and ΔCq= Cqcontrol- Cqsample. Data from both runs were used in the fold change calculation. Data was graphed using R statistical software (Figure 2).17 Differences in gene expression were analyzed for significance using Bootstratio. Bootstratio is an application which does not assume an underlying probability distribution and uses resampling methods to look at the significance of relative gene expression levels determined by qPCR.18
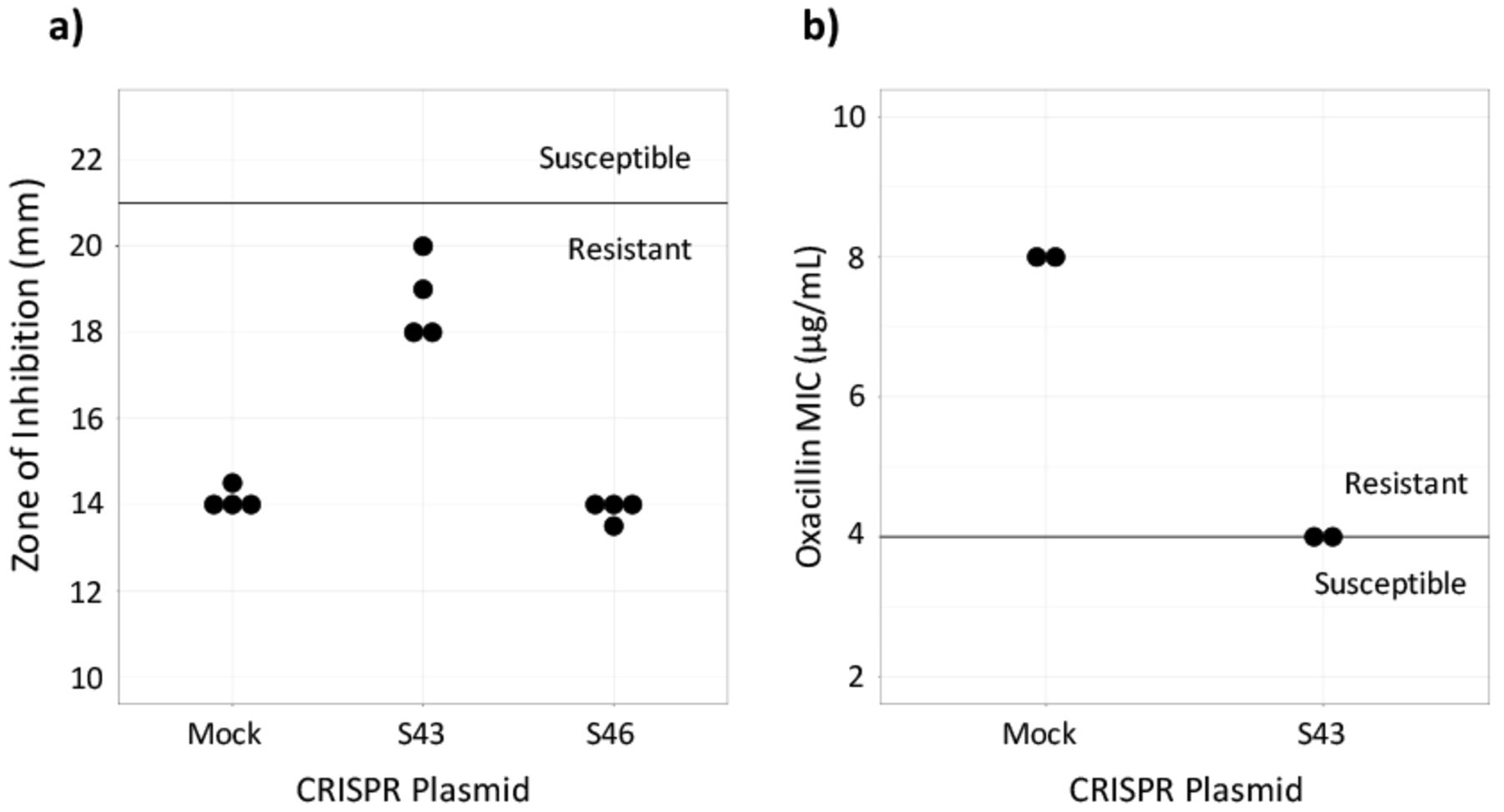
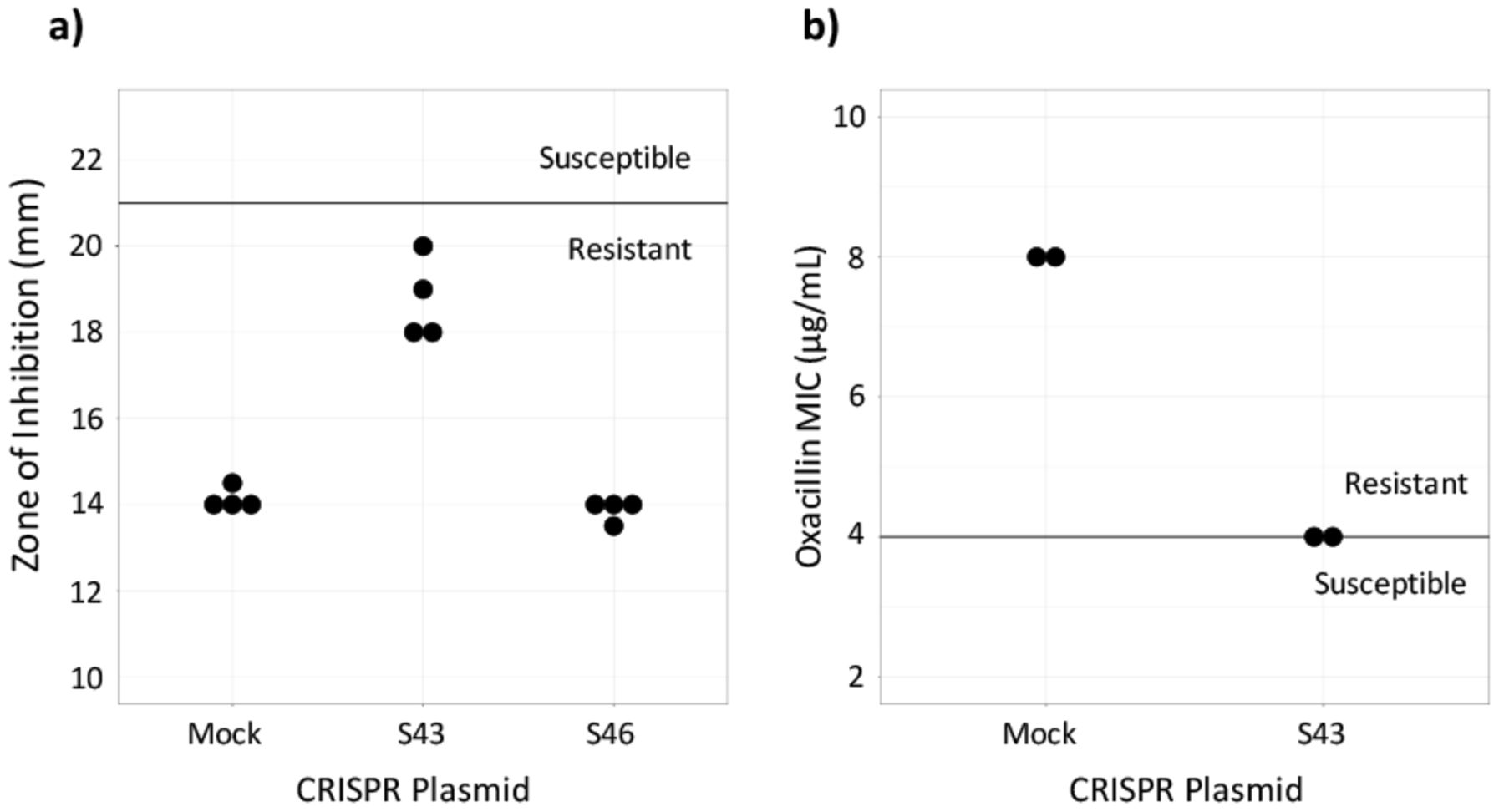
Susceptibility testing on CRISPR-dCas9 treated isolates. a) Cefoxitin disk diffusion testing was performed in quadruplicate. The dark line represents the breakpoint between resistant and susceptible isolates. b) Oxacillin Microbroth serial dilutions performed in duplicate confirmed results from the disk diffusion test. Plasmid S43 reduced cefoxitin resistance, but did not achieve in vitro susceptibility and plasmid S46 had no effect (data not shown).
RESULTS
MIC Testing
To assess if the CRISPR-dCas9 system induced in vitro susceptibility, the MIC of test and control MRSA batches were found. This was done by performing an oxacillin disk diffusion test. Industry standards provided by the CLSI require zones of inhibition greater than or equal to 22 mm for MRSA to be susceptible to cefoxitin and zones less than or equal to 21 mm for the MRSA to be resistant to cefoxitin.19 Results showed that the control, MRSA ATCC 43300 transformed with the mock plasmid, was resistant to beta-lactam antibiotics. It had an average zone of inhibition of 14.13 mm. MRSA with the S46 plasmid displayed zones of inhibition similar to the control, with an average diameter of 13.86. Plates of MRSA with the S43 plasmid had an average zone of inhibition of 18.75 mm. The S43 group was not susceptible to cefoxitin, but it had a wider zone of inhibition compared to the control (Figure 2a). Because the S46-transformed MRSA did not largely affect the bacteria, only the S43 and control bacteria were used in continued testing.
The CLSI's breakpoints for oxacillin susceptibility testing through Microbroth serial dilutions are an MIC of less than or equal to 2 μg/mL to be susceptible and an MIC of greater than or equal to 4 μg/mL to be resistant.19 The MIC for the S43-transformed bacteria as determined by oxacillin serial dilutions was 4 μg/mL and the MIC for the mock-transformed control bacteria was 8 μg/mL (Figure 2b). This confirmed the results of the disk diffusion test, showing that the S43 plasmid had caused the MRSA to become less antibiotic resistant, though not entirely susceptible.
mecA Expression Analysis
After observing a partial reduction in beta-lactam antibiotic resistance during antibiotic testing, RT-qPCR was performed to analyze mecA gene expression levels. RT-qPCR runs yielded consistent amplification and smooth melt curves (Figure 1), showing that only desired targets were amplified. Two independent runs were performed, showing an average fold difference of 0.230, showing that the S43 plasmid significantly reduced (p < 0.0005) mecA gene expression by 77% as compared to the mock group (Figure 3). The average calculated PCR efficiencies for the 16s and mecA samples were 67.6% and 75.8%, respectively. These results showed that the CRISPR-dCas9 system had an incomplete effect in suppression of mecA mRNA expression.
DISCUSSION
Two CRISPR-dCas9 systems were designed to suppress antibiotic resistance in MRSA bacteria by targeting the mecA methicillin resistance gene. The S43 plasmid targeted the coding strand of mecA and the S46 plasmid targeted the noncoding strand. MRSA bacteria with the S46 plasmid and MRSA with the S43 plasmid were still resistant to beta-lactam antibiotics, though S43 batches were less resistant, as shown by cefoxitin disk diffusion tests and oxacillin microbroth serial dilutions. RT-qPCR showed that the S43 plasmid reduced mecA gene expression in MRSA to 0.230 of its original value, representing a 77% reduction. Although mecA gene expression in MRSA was significantly reduced (p<0.0005) by the S43 CRISPR-dCas9 system, MIC testing showed that this decrease was not enough to make MRSA fully susceptible to beta-lactam antibiotics by in vitro standards. Previous studies have been successful in using CRISPR-Cas9 systems to genetically modify MRSA, which resulted in killing the bacteria. This is the first study to use CRISPR-dCas9 systems in S. aureus, which succeeded in reducing beta-lactam antibiotic resistance while maintaining bacterial life. The CRISPR-dCas9 system studied here may be a viable option for treatment of antibiotic-resistant bacteria because it does not kill the target bacteria, as opposed to the CRISPR-Cas9 system used in other studies.
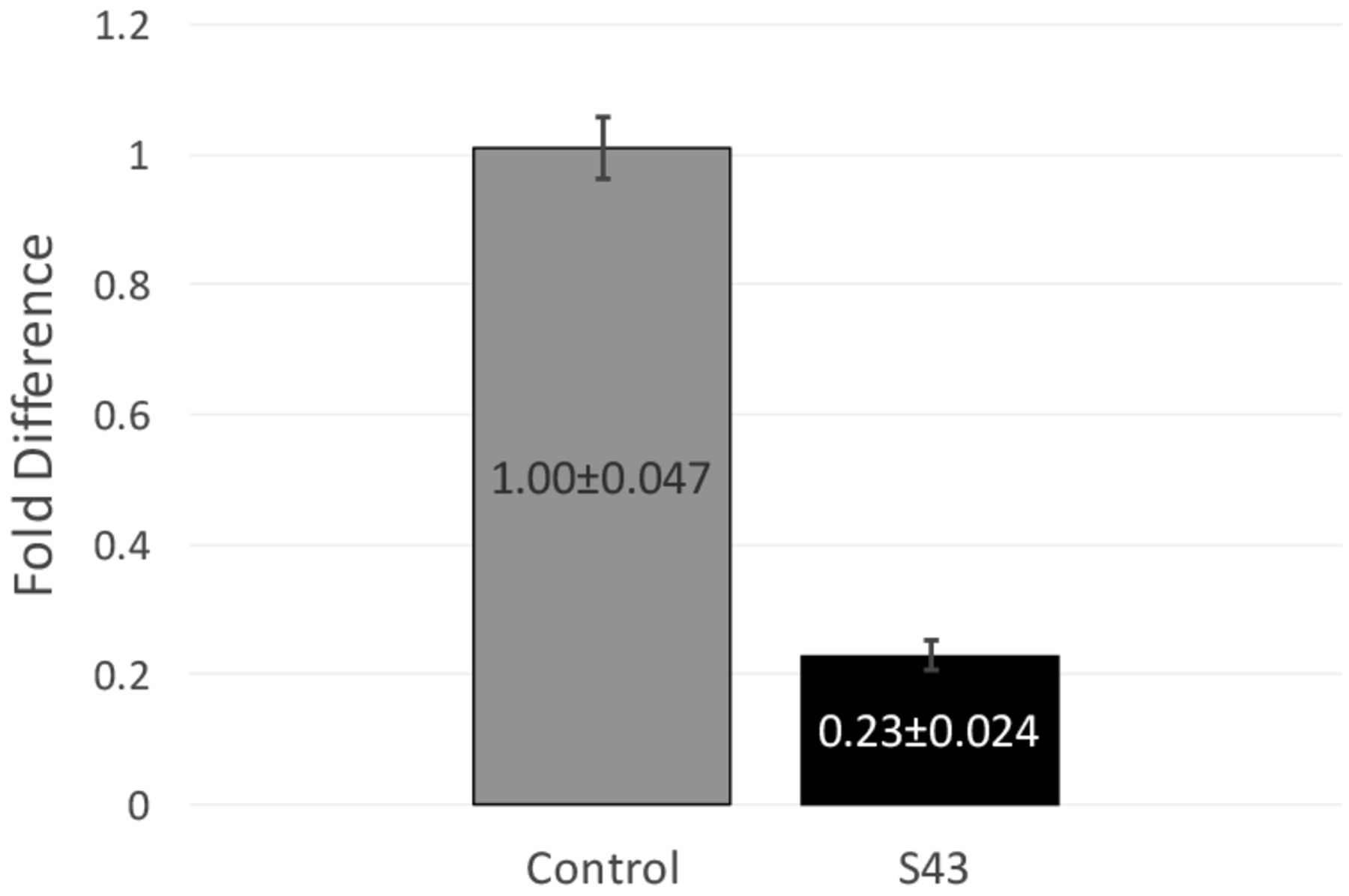
CRISPR-dCas9 system significantly decreased mecA expression in MRSA. mecA mRNA expression was significantly reduced in S43 CRISPR-dCas9 transformed MRSA (p<0.0005). mecA expression was reduced by 77% to 0.23 of its original value, as compared to a mock-transformed control. Fold differences were calculated using the relative standard curve method. Shown above are fold differences ± standard error of the mean.
This study could be enhanced by testing the S43 and S46 plasmids for synergistic effects. This would be accomplished by co-transforming the plasmids into the same organisms, then performing the MIC testing and mecA expression analysis. Different target sites on the mecA gene could also be explored. RT-qPCR allows for analysis of gene expression levels, but gene to protein expression ratios are not always 1:1. For this reason, a western blot test should be completed to look at PBP 2A protein expression differences in S43-transformed MRSA versus expression in the control. The intention behind keeping the bacteria alive by using the CRISPR-dCas9 is to introduce into the environment a self-propelling CRISPR-dCas9 system to suppress mecA. This study succeeded in keeping the bacteria alive, and further attempts could be made to choose a target site which fully suppresses the mecA gene. After the CRISPR-dCas9 plasmid system is fully optimized, it could be integrated onto the bacterial chromosome in order to become self-sustaining. This may be done through bacterial transformation. Small-scale testing would follow to determine whether the CRISPR-dCas9 system is effectively passed to offspring each time the MRSA reproduce. This same concept can be applied to the plethora of other antibiotic-resistant bacteria. One CRISPR-dCas9 system is capable of housing multiple target sites, meaning that the same plasmid may be used to target multiple resistance mechanisms in the same organism, or resistance in different types of bacteria. Eliminating the drug resistance of bacteria could wipe out their standing as health threats. Traditional antibiotics could once again be used to effectively treat these bacteria.
ACKNOWLEDGEMENTS
The authors thank Dr. David Bikard for his advice regarding this project and the University of Utah CRISPR Core Facility for producing the CRISPR plasmids used in this experiment.
Financial support was provided by The Signpost at Weber State University, Utah STEM Action Center, Ritchey Science Fair Fund, and Weber State University Medical Lab Sciences Department.
- © Copyright 2018 American Society for Clinical Laboratory Science Inc. All rights reserved.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Harnessing CRISPR interference to re-sensitize laboratory strains and clinical isolates to last resort antibiotics
- Precision Genome Editing Unveils a Breakthrough in Reversing Antibiotic Resistance: CRISPR/Cas9 Targeting of Multi-Drug Resistance Genes in Methicillin-Resistant Staphylococcus aureus
- CRISPR Interference for Rapid Knockdown of Essential Cell Cycle Genes in Lactobacillus plantarum