


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Tarleton State University
- Tarleton State University
- Tarleton State University
- Tarleton State University
- Address for Correspondence: Sara Taylor
, Tarleton State University, sataylor{at}tarleton.edu
ABSTRACT
The patient was a 33-year-old woman at 31 weeks gestation with twins who presented to the emergency department complaining of shortness of breath, headache, and blurry vision. The patient’s preliminary complete blood count, red blood cell morphology, coagulation testing, and certain metabolic indicators were characteristic of a hemolytic process caused by microcirculatory lesions known as thrombotic microangiopathies. The major pathologies of this hemolytic process are thrombotic thrombocytopenic purpura, hemolytic uremic syndrome (HUS), disseminated intravascular coagulation, and hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome. Additional coagulation and biochemical testing indicated that the patient probably was experiencing HELLP syndrome, but atypical HUS (aHUS) could not be ruled out. Consequently, an aHUS genetic susceptibility panel was also ordered for this patient. The results of the genetic testing revealed that the patient did indeed have aHUS, a disease of complement dysregulation. In approximately 50% of patients, mutations have been described in the genes that encode complement regulator factors. With an accurate diagnosis established, the patient was able to receive treatment using an anti-C5 monoclonal antibody aimed specifically at controlling the dysregulated complement protein C5.
- aHUS - atypical hemolytic uremic syndrome
- ADAMTS13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif - member 13
- ALT - alanine aminotransferase
- AST - aspartate aminotransferase
- APTT - activated partial thromboplastin
- BNP - B-type natriuretic peptide
- CBC - complete blood count
- CFH - complement factor H
- CK-MB - creatine kinase-muscle/brain
- DIC - disseminated intravascular coagulation
- FDP - fibrin degradation product
- fH - factor H
- HELLP - hemolysis - elevated liver enzymes - and low platelet count
- HUS - hemolytic uremic syndrome
- INR - international normalized ratio
- LDH - lactate dehydrogenase
- RBC - red blood cell
- SUN - serum urea nitrogen
- TMA - thrombotic microangiopathy
- TTP - thrombotic thrombocytopenic purpura
- WBC - white blood cell
CASE REPORT
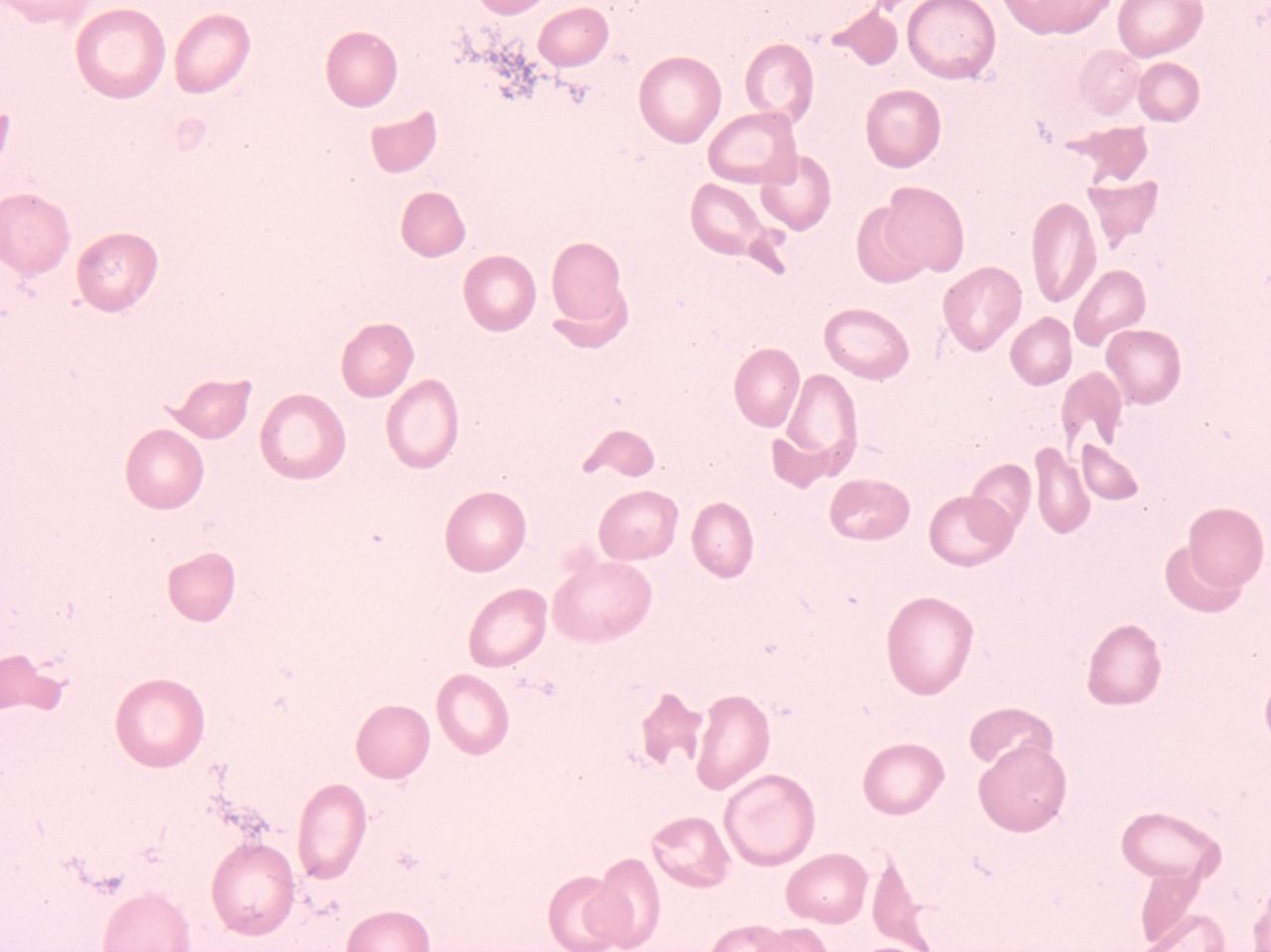
A 33-year-old woman at 31 weeks gestation with twins presented to the emergency department complaining of shortness of breath, headache, and blurry vision in her left eye. Her pregnancy to date was without complications except for significant edema and, very recently, a urinary tract infection being managed with amoxicillin. Her blood pressure was 158/98. Her inaugural hematology workup showed her to have a markedly increased white blood cell (WBC) count while displaying neutrophilia, anemia, and thrombocytopenia (Figure 1, Table 1). A manual leukocyte differential performed at this time revealed the presence of schistocytes, although the numbers were modest (Figure 1, Table 1). The patient had normal results for prothrombin time, activated partial thromboplastin (APTT) times, and fibrinogen levels, but her fibrin degradation products (FDPs) were modestly increased. Significantly abnormal chemistry values found included a moderately elevated serum urea nitrogen (SUN), elevated creatinine, and mildly to markedly elevated liver enzymes (alanine aminotransferase [ALT], aspartate aminotransferase [AST], and lactate dehydrogenase [LDH]) (Table 1). A urinalysis done at this time revealed the presence of blood, leukocytes, protein, bacteria, and yeast. A urine culture and sensitivity done at the time of admission could not be interpreted because of specimen contamination (data not shown).
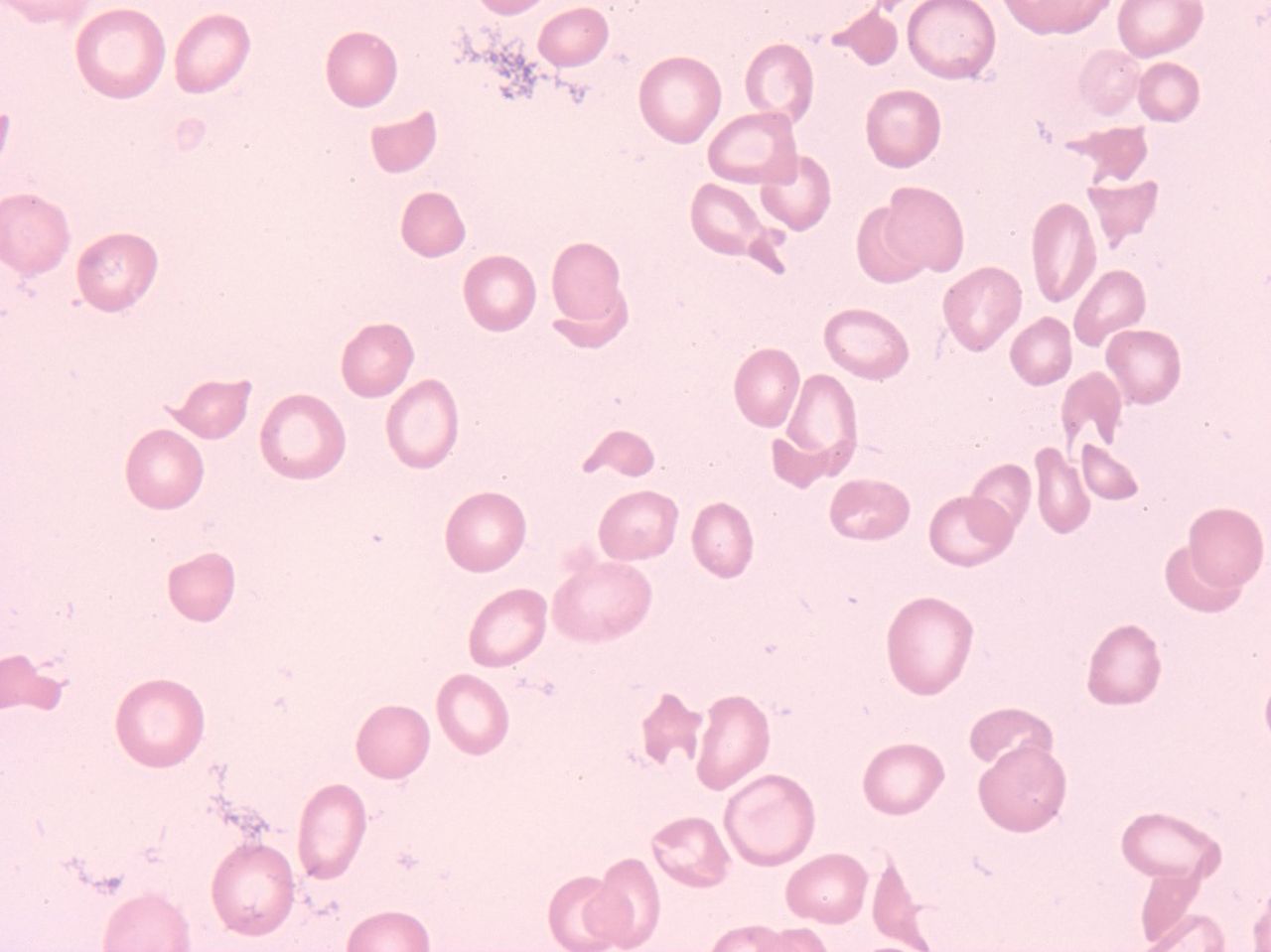
Peripheral blood displaying a significantly low platelet count (42,000/uL) and shistocyes. 100X magnification.
Diagnostic timeline
LABORATORY FINDINGS
The patient’s preliminary complete blood count (CBC), red blood cell (RBC) morphology, coagulation testing, and certain metabolic indicators were characteristic of a hemolytic process caused by microcirculatory lesions known as thrombotic microangiopathies (TMAs).1,2 Laboratory testing proceeded so that the major pathologies of this hemolytic process could be ruled out. Hemolysis in TMAs is caused by damage to the endothelial lining of the smallest blood vessels, the damage activates the coagulation cascade, and the resulting fibrin strands fragment erythrocytes caught in the fibrin structure.3 Several diseases underlie the development of TMA, including antiphospholipid antibody syndrome, disseminated intravascular coagulation (DIC), malignant hypertension, thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), atypical HUS (aHUS), and a severe form of preeclampsia known as hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome.3⇓-5 The patient’s normal international normalized ratio (INR) and APTT test results indicated that her TMA was not the result of antiphospholipid antibodies.6 These same coagulation results, along with a normal fibrinogen level and only slightly elevated FDPs concomitant with a nonsupportive clinical presentation, allowed us to rule out DIC for this patient as well.6,7
Malignant hypertension remained a possibility because the patient presented to our facility pregnant, with a headache, and with blurry vision.8,9 To investigate the possibility of this disorder, creatine kinase-muscle/brain (CK-MB) levels, troponin I testing, and a careful eye examination were carried out. Both cardiac markers indicated that the patient had a low probability of cardiac damage, and she tested negative for burry vision, diplopia, scotoma, photophobia, coryza, and oculorrhea (data not shown). Considering the results of her cardiac markers and her visual test results, it was determined that pathologies other than malignant hypertension be considered as the likely cause of her TMA.8,9 To investigate the possibility of TTP, a blood sample was sent to a reference laboratory to be tested for a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity.5,6,10 The result of the ADAMTS13 activity assay test was charted 5 days after the patient was admitted. Reduced activity of ADAMTS13 (INR > 61%) indicated that the cause of our patient’s hematology troubles could be complicated by TTP, although her ADAMTS13 activity was not as critically low as is usually seen in classic TTP.5,6,10 Accordingly, the patient began plasma exchange therapy. At the same time that ADAMTS13 activity was investigated, testing for the presence of Shiga toxin 1 and 2 was carried out to rule out HUS.5,6,11 Negative test results for the presence of Shiga toxins supported a conclusion that this patient’s hemolytic troubles were not caused by HUS, but there remained the possibility that our patient’s trouble was a variant of HUS–aHUS. This prospect was investigated with the assessment of complement proteins. The results of complement testing done on day 5 revealed normal results for both C3 and C4, which indicated that the underlying problem was not likely aHUS because C3 levels would be expected to be decreased in this pathology.5 The patient’s ongoing SUN and creatinine levels indicated that she was experiencing renal failure, so a renal biopsy was done on day 10, a few days after a caesarian section was performed and twins were delivered. Changes consistent with toxemia of pregnancy with progression to frank TMA were noted on the biopsy specimen (Table 1). Given the history of recent pregnancy, these tissue findings most likely represented HELLP syndrome, although it was noted that aHUS could not be ruled out. On day 10, the patient’s B-type natriuretic peptide (BNP) was interrogated to clarify whether or not her difficulty was indeed HELLP syndrome.12 As can be seen in Table 1, our patient’s BNP strongly supported our early suspicion that the patient had HELLP syndrome, again with TTP complicating her troubles.12 However, to fully investigate the possibility of aHUS, an aHUS genetic susceptibility panel was also ordered. This multigene panel interrogates pathogenic variants in the genes that are associated with genetic aHUS (more discussion to follow).13 When the results of the genetic susceptibility panel were received, they indicated that a diagnosis of aHUS was a strong candidate for this patient because one of her alleles contained a CFHR3–CFHR1 deletion (Table 2). At this point, the patient’s therapy was changed so that she began receiving eculizumab, a monoclonal antibody that is effective in the management of aHUS.7,14 The patient’s problems rapidly resolved, confirming aHUS as the cause of her troubles.
aHUS susceptibility panel
DISCUSSION
The complement system of proteins is part of the innate immune system.3,4 Activation of the complement cascade of proteins occurs by 1 of 3 pathways: classical, lectin, and the alternative pathway. All 3 pathways produce an enzyme that is active midway in the complement cascade: C3 convertase. C3 convertase activates a C5 that, in turn, may activate the terminal portion of the complement cascade.3⇓-5 Once it is fully activated, the complement cascade must be tightly regulated to avoid cell damage.3,9 Eculizumab is an anti-C5 monoclonal antibody that specifically targets dysregulated C5; thus, it regulates an important complement protein that is active midway through the complement cascade.7,14
The specific etiology of aHUS appears to be dysregulated C3 convertase activity.15⇓-17 Whereas faulty C3 protein itself accounts for a small number of aHUS cases (~5%), there are a number of additional complement cascade components that appear to underlie the development of aHUS when the results are abnormal.13,15⇓-17
Complement factor H glycoprotein (fH) coded by complement factor H (CFH) is a major regulator of complement activity.18 Located in close proximity to CFH on the long arm of chromosome 1, there are 5 genes that code for proteins that appear to control the activity of fH. These genes are known as CFH-related genes; they are designated as CFHR1-5.13,15⇓-17 The protein products of these fH-related genes show immunological cross-reactivity with one another and with fH as well.16 Rearrangements in the CFH-CFHR1-5 gene cluster can result in several pathologies; CFHR1 and CFHR3 mutations are especially implicated in the development of aHUS.13,15⇓-17 CFHR1 and CFHR3 mutations are common, and they increase one’s risk of developing aHUS because these mutations appear to increase the likelihood of developing antibodies to the regulatory fH.15⇓⇓-18 If these autoantibodies develop, a loss of complement control is likely. In addition to the fH-related proteins, 5 additional complement proteins appear to contribute to the development of aHUS when their function is deviant. Complement factors B, H, and I; membrane cofactor protein; and thrombomodulin are coded for by CFB, CFH, CFI, MCP, and THBD genes.13 Mutations in fH-related genes and these specific complement protein genes collectively are believed to underlie nearly 50% of aHUS cases.13
SUMMARY/CONCLUSION
TMAs are hemolytic conditions caused by microcirculatory lesions. Diseases that lead to the development of TMA include antiphospholipid syndrome, DIC, malignant hypertension, HUS, aHUS, TTP, and a severe form of preeclampsia known as HELLP syndrome. Our patient’s inaugural CBC, SUN/creatinine levels, and liver enzymes suggested that she was experiencing a TMA that is characterized by compromised RBC and platelet parameters, compromised kidney function, and elevated liver enzymes. Further laboratory testing was directed towards diagnosing the exact nature of our patient’s problem. Accordingly, additional coagulation tests, vision testing, a urine culture and sensitivity test (data not shown), Shiga toxin testing, C3 and C4 testing, a renal procedure, an ADAMTS13 activity assay, and BNP were ordered to clarify the exact nature of this patient’s TMA. The results of these tests did not offer a clear-cut interpretation (see Laboratory Findings), but, considering the patient’s clinical history of a current pregnancy, it was decided that the patient most likely had HELLP syndrome, possibly complicated by TTP. The patient was started on plasma exchange therapy, although her ADAMTS13 activity was not as low as is generally seen in classic TTP. After a week of plasma exchange, the patient’s laboratory values did not improve satisfactorily, and she was still experiencing disturbing clinical symptoms. At this time, an aHUS susceptibility panel was ordered on our patient with results that suggested that her problem was aHUS, although the typical decreased levels of C3 were not present. The patient’s therapy was changed to a regimen of eculizumab, and her problems were resolved. This outcome was supportive of a diagnosis of aHUS.
- Received July 7, 2018.
- Accepted January 9, 2019.
American Society for Clinical Laboratory Science
References




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- aHUS - atypical hemolytic uremic syndrome
- ADAMTS13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif - member 13
- ALT - alanine aminotransferase
- AST - aspartate aminotransferase
- APTT - activated partial thromboplastin
- BNP - B-type natriuretic peptide
- CBC - complete blood count
- CFH - complement factor H
- CK-MB - creatine kinase-muscle/brain
- DIC - disseminated intravascular coagulation
- FDP - fibrin degradation product
- fH - factor H
- HELLP - hemolysis - elevated liver enzymes - and low platelet count
- HUS - hemolytic uremic syndrome
- INR - international normalized ratio
- LDH - lactate dehydrogenase
- RBC - red blood cell
- SUN - serum urea nitrogen
- TMA - thrombotic microangiopathy
- TTP - thrombotic thrombocytopenic purpura
- WBC - white blood cell