


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Larry Smith
, Abbott Diagnostics–Hematology, larry.smith{at}abbott.com
LEARNING OBJECTIVES
1. Discuss the pathogenesis of acquired and congenital thrombotic thrombocytopenic purpura (TTP).
2. Outline the function of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13.
3. Describe the role of neutralizing antibodies in TTP.
4. Correlate laboratory findings from the coagulation laboratory related to TTP.
5. List treatment options available for treating patients with TTP.
ABSTRACT
This review describes classic thrombotic thrombocytopenic purpura (TTP), discusses the pathogenesis of acquired and congenital TTP, describes clinical and laboratory manifestations observed in patients, and lists options for treating patients with TTP. TTP is a rare hematologic disorder characterized by thrombocytopenia and microangiopathic hemolytic anemia. It results from a congenital or acquired deficiency of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13), in plasma. Most cases are caused by an autoimmune mechanism that interferes with ADAMTS-13; however, rare inherited forms of TTP have been described (e.g., Upshaw-Schulman syndrome). It is still considered a life-threatening disease with a mortality rate of 10%–20%. Severe deficiency of ADAMTS-13 (<10%) is most often associated with congenital TTP. Although TTP is a serious hematologic emergency that is almost always fatal in untreated cases, an understanding of its pathophysiology can lead to successful treatment strategies, resulting in improved patient care and outcomes.
- ADAMTS-13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif - member 13
- aPTT - activated partial thromboplastin time
- BU - Bethesda unit
- EC - endothelial cell
- FDA - Food and Drug Administration
- FIB - fibrinogen
- GPIb - glycoprotein Ib
- HUS - hemolytic uremic syndrome
- IgG - immunoglobulin G
- MAHA - microangiopathic hemolytic anemia
- NAC - N-acetylcysteine
- PT - prothrombin time
- rADA - recombinant ADAMTS-13
- TMA - thrombotic microangiopathy
- TPE - therapeutic plasma exchange
- TTP - thrombotic thrombocytopenic purpura
- ULVWF - unusually large von Willebrand factor
- USS - Upshaw-Schulman syndrome
- VWF - von Willebrand factor
- VWF-CP - von Willebrand factor–cleaving protease
- WPB - Weibel-Palade body
- ADAMTS-13
- hemolytic uremic syndrome
- microangiopathic hemolytic anemia
- thrombotic microangiopathies
- thrombotic thrombocytopenic purpura
- von Willebrand factor-cleaving protease
- Upshaw-Schulman syndrome
INTRODUCTION
Thrombotic thrombocytopenic purpura (TTP) is a rare, life-threatening disease that is considered a medical emergency with a very high mortality rate (90%) if not managed in a timely manner.1 It is one of a group of disorders known as the thrombotic microangiopathies (TMAs), which includes TTP, hemolytic uremic syndrome (HUS), and disseminated intravascular coagulation. It is characterized by a pentad of features, including thrombocytopenia, microangiopathic hemolytic anemias (MAHAs), neurological deficits, renal dysfunction, and fever (Box 1).2
The estimated incidence of TTP ranges from 3–11 cases per million residents per year.3 It was first described in 1924 by Eli Moschowitz in a 16-year-old female who presented with weakness in the upper extremities and a few petechiae on the left arm and later died of a fatal TMA.4,5 Postmortem microscopic examination revealed widespread hyaline thrombi deposited in capillaries and arterioles.5
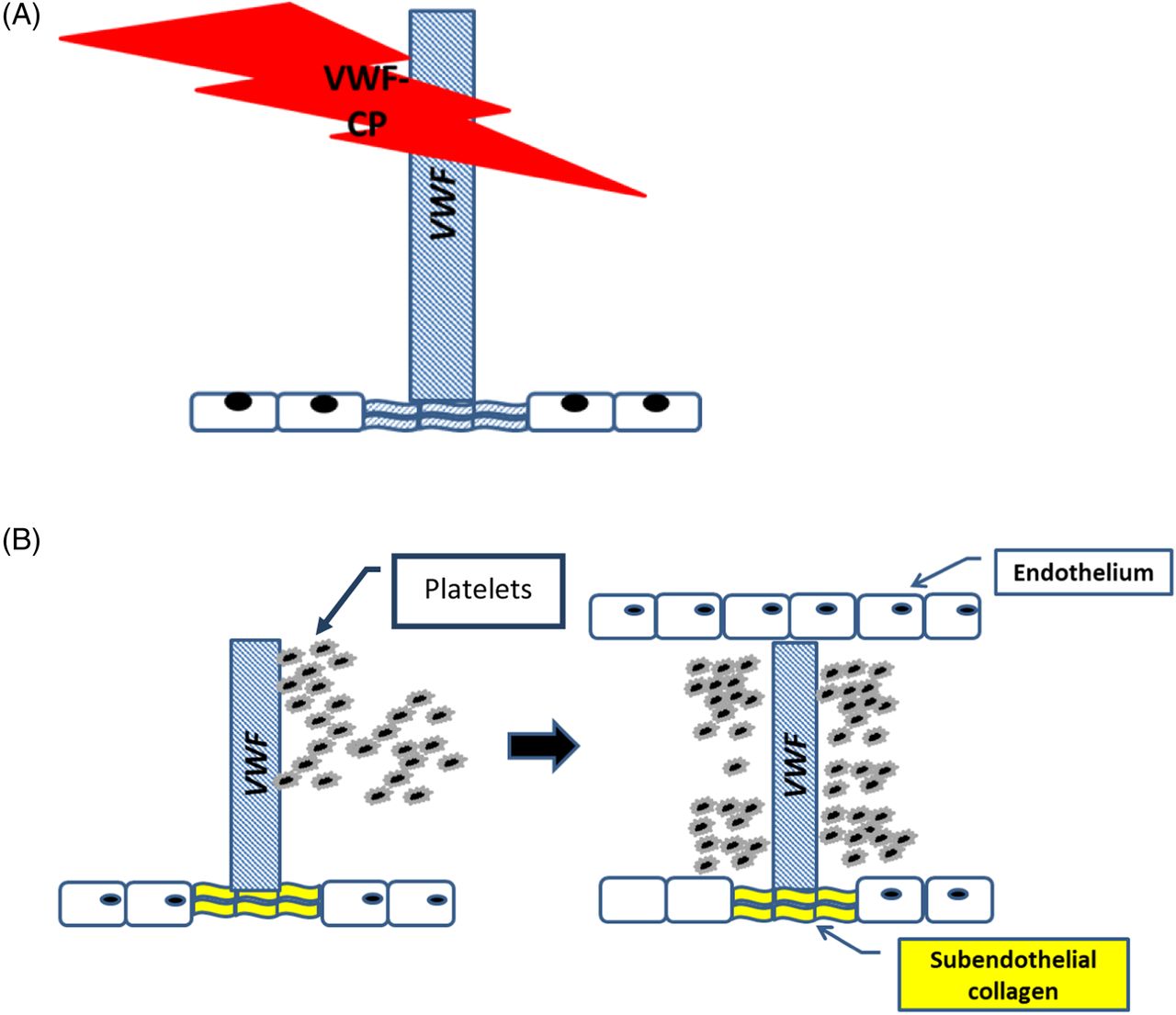
Prior to the 1980s, the cause of TTP was relatively unknown, and the mortality rate was greater than 90%. It was later shown that a substance in plasma could reduce the reoccurrence of relapsing episodes of chronic TTP.6 In 1982, von Willebrand factor (VWF), a multimeric glycoprotein that plays a critical role in platelet adhesion and platelet aggregation at high shear rate was associated with TTP.4 Moake et al reported on the presence of unusually large von Willebrand factors (ULVWFs) in the plasma of patients with chronic relapsing TTP.7,8 He proposed that the ULVWFs were responsible for the hyperactive platelet adhesion and aggregation observed in these patients. Immunohistochemical studies later confirmed the presence of ULVWF in the platelet microthrombi.9 It was suggested that a protein capable of regulating VWF multimer size was missing, defective, or inhibited. This protein was later identified as von Willebrand factor–cleaving protease (VWF-CP),10,11 also known as a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13) (Figure 1A and B).12

ADAMTS-13 (also known as VWF-CP). (A) Normal VWF-CP cleaving ULVWF cleaves the ULVWF into smaller multimers that have decreased platelet-binding affinity, whereas (B) absent VWF-CP does not cleave ULVWF that has increased platelet-binding affinity and vessel occlusion.
TTP can occur as a congenital disease or an acquired disease. Congenital TTP, also known as Upshaw-Schulman syndrome (USS), is a rare disorder that is characterized by single or recurrent episodes of thrombocytopenia, MAHA, and microvascular thrombosis that lead to ischemia in multiple organs.13 It is caused by a mutation in the ADAMTS-13 gene and prevents the production of the ADAMTS-13 metalloprotease. It has an autosomal recessive mode of inheritance. More than 70 genetic mutations of the ADAMTS-13 gene have been described.1 A severe deficiency of ADAMTS-13 protease results in persistence of the ULVWF multimers in plasma. The uncleaved ULVWF multimers in the microcirculation cause the formation of platelet-rich thrombi that are responsible for fragmented erythrocytes in the circulation, one of the hallmarks of the TMAs. About 5% of all TTP cases with a deficiency of ADAMTS-13 are congenital.1 Patients present with symptoms at a very early age (around 5 years of age) and often experience relapsing episodes of TTP throughout life.
The most common form of TTP is acquired; incidences peak between ages 30 and 50. Acquired TTP is more prevalent in women than men. It can present as primary or secondary TTP. Primary TTP (idiopathic) is associated with severely decreased ADAMTS-13, whereas secondary TTP results from a variety of conditions and has a poorer prognosis than primary TTP.14 Autoantibodies (e.g., immunoglobulin G [IgG]) are directed against the enzyme activity of the protease, reducing its ability to cleave the ULVWF multimers.15 TTP has also been associated with other conditions, such as pregnancy, malignancy, and transplantation, and some drugs.
PATHOGENESIS OF TTP
TTP involves the formation of microcvascular thrombi in arterioles, capillaries, and many organs. The microvascular thrombi are composed of platelet aggregates primarily with very little fibrin. In addition, there is neither perivascular inflammation nor overt endothelial cell (EC) damage.15 This results in decreased blood flow to vital organs, such as the brain, heart, and kidney.
ADAMTS-13 is a metalloprotease found in plasma that is responsible for cleaving VWF in a shear dependent manner.16,17 It is secreted from ECs in the vascular lining of blood vessels. It plays a critical role in cleaving the unusually large multimers of VWF in plasma. The gene for ADAMTS-13 is located on chromosome 9q34.
VWF is a large adhesive protein that is synthesized in ECs and megakaryocytes. It is stored in the Weibel-Palade bodies (WPBs) of ECs and in the α granules of megakaryocytes. It is composed of multimeric units that range in size up to greater than 20 million Da. Because of its large size, it is tightly coiled and packaged in the WPBs. When VWF is released into plasma from damaged ECs, it binds to the glycoprotein Ib (GPIb) receptor on circulating platelets. The bound platelet is pulled down to the damaged endothelial surface, where it binds or adheres. As the adherent platelet becomes activated, it attracts additional platelets, leading to platelet aggregation. The ULVWF multimers are very thrombogenic. They have a very strong binding affinity for the GPIb receptors on circulating platelets. In addition, some of the ULVWF multimers remain attached to the damaged endothelial surface, where they are capable of self-association to form thick fibers.18 The thick fibers containing surface-bound ULVWF are also very thrombogenic. They spontaneously bind to platelets, creating the beads-on-a-string image. Functional ADAMTS-13 is responsible for cleaving VWF into smaller multimers, which reduce the spontaneous binding to platelets and thrombogenicity.
CLINICAL AND LABORATORY FEATURES
Patients with TTP often present with a constellation of features. Symptoms are similar to those seen in other TMAs and are characterized by microvascular thrombosis, anemia, and associated organ dysfunction. The hallmark feature is fragmentation of erythrocytes by the microvascular thrombi present in the associated anemia. TTP is associated with a pentad of symptoms, including thrombocytopenia (70%–100%), MAHA (70%–100%), neurologic deficits (50%–90%), renal deficits (50%), and fever (25%).14 Waiting for the entire pentad of features to be present before arriving at a diagnosis of TTP could have severe clinical consequences. Thrombocytopenia and MAHA in the absence of the remaining symptoms are sufficient to suspect TTP.
A complete blood cell count serves to highlight the anemia and thrombocytopenia with a decrease in hemoglobin and thrombocytopenia. The reticulocyte count, red cell distribution width, and mean platelet volume are often increased. The peripheral blood smear contains fragmented red blood cells (schistocytes). The routine coagulation assays, such as the prothrombin time (PT), activated partial thromboplastin time (aPTT), and fibrinogen (FIB), are within normal limits (Table 1). Assays for VWF activity, VWF antigen, ristocetin cofactor, and factor VIII are typically normal, whereas VWF multimer analysis shows an increase in the ULVWF multimers. Several specific assays for detecting and measuring ADAMTS-13 are available and include activity assays, antigen assays, and inhibitor assays (for neutralizing and nonneutralizing antibodies).
Admission laboratory values for a 40-year-old female who was seen in the emergency department with abdominal pain over 3 days
The laboratory values for a 40-year-old-female who was seen in the emergency department with abdominal pain over 3 days are included in Table 1. The results of the ADAMTS-13 activity assay and the inhibitor assay were decreased: ADAMTS-13 activity, <5%; ADAMTS-13 inhibitor titer, 2 Bethesda units (BU) (Table 1). These findings, a severely decreased ADMTS-13 activity level and the presence of an inhibitor, are consistent with acquired TTP. A severe decrease in ADAMTS-13 activity in the absence of an inhibitor should raise clinical suspicion for congenital TTP.19
Laboratory evaluation will be discussed in this review, but it is by no means a complete listing of all of the assays available for detecting and measuring ADAMTS-13. VWF activity assays involve adding patient plasma to a solution containing VWF and measuring ADAMTS-13–cleavage products.19 The VWF used in the solution component for these assays contains degradation products of VWF multimers (purified, plasma derived, or recombinant) or VWF peptides (synthetic).20 Several different principles are used to measure the cleavage products previously mentioned, either directly or indirectly. These include electrophoresis, platelet aggregometry, fluorescence resonance energy transfer, and immunoassay. The assays based on VWF multimeric detection are sensitive down to 3%–6% of ADAMTS-13 activity, whereas assays based on VWF peptide detection are sensitive down to 1%–3% of ADAMTS-13 activity.20 Plasma ADAMTS-13 activity levels range from 50% to 180% in individuals with normal results, whereas reduced levels can occur in a number of clinical conditions, such as liver disease, malignant disease, inflammatory disorders, and pregnancy. Activity levels less than 10% are associated with acute TTP.1 It is important to recognize that approximately two-thirds of patients with a clinical diagnosis of TTP will have ADAMTS-13 activity levels less than 10%.14
Immunoassays are available for measuring ADAMTS-13 antigen. These assays use either monoclonal or polyclonal antibodies. Several kits are available; however, they may vary in their ability to discriminate among full-length, mutant, and truncated forms of ADAMTS-13 protein.20 It is also not known how well these kits function in the presence of autoantibodies and immune complexes.20 There are 2 types of antibodies directed at ADAMTS-13. Neutralizing antibodies (anti–ADAMTS-13 inhibiting antibodies) are circulating autoantibodies that inhibit the activity of the VWF-CP. They can be detected using classic mixing studies employing various dilutions (1:1, etc) of heat-inactivated patient plasma and normal plasma. Neutralizing antibodies are present in most cases of acquired, idiopathic TTP. The second type of antibody is a binding antibody (nonneutralizing anti–ADAMTS-13 antibody). It binds to the VWF-CP and accelerates its clearance from the circulation. They are found in 10%–15% of patients with severe deficiency of ADAMTS-13. They are usually IgG or immunoglobulin M autoantibodies.14 They can be detected using enzyme-linked immunosorbent assays that are commercially available. It is imperative for laboratories to validate the assay in-house because of the heterogeneity of the substrate VWF and the differences in endpoint detection of the different assay principles available. Results across different methodologies may not be interchangeable, so the same method used for initial diagnosis should be used for follow-up in patient care.
PATIENT CARE
Because TTP is considered a medical emergency, a clinician with a high index of suspicion should initiate therapy immediately. The original clinical description of TTP involves the classic pentad of features discussed early in this review. It has been shown that this pentad is neither sensitive nor specific, and the majority of patients presenting with TTP do not have all 5 clinical features.19 TTP should be suspected in the presence of MAHA and thrombocytopenia alone.19 A number of treatment options are currently available for treating patients with TTP (Table 2). These include therapeutic plasma exchange (TPE) therapy; immunomodulatory therapies, including corticosteroids and Rituximab; and novel targeted therapies, including Caplacizumab, N-acetylcysteine (NAC), Bortezomib, recombinant ADAMTS-13 (rADA), and Anfibatide. At the time of this publication, some of these treatments are approved by the Food and Drug Administration (FDA) for treating patients with TTP, and some are still in clinical trial phase, which are FDA-approved. TPE is the mainstay of management of acute TTP. It replenishes ADAMT-13 activity and removes anti–ADAMTS-13 antibodies, ADAMTS-13 immune complexes, and ULVWF.19 All other treatments are described in Table 2. Rituxamab is a chimeric monoclonal antibody directed at CD20 on mature B lymphocytes downregulating the immune response.19 Caplacizumab is a bivalent humanized nanobody that attaches to the A1 domain of VWF and inhibits its interaction with the GPIb receptor on platelets, reducing their activity.19 NAC is an adjuvant therapy with a structure similar to disulfide linkages in VWF used to reduce large VWF multimers and inhibits VWF-dependent platelet aggregation.19 Bortezomib is a proteasome inhibitor that inhibits B-lymphocyte antibody production. rADA replaces circulating ADAMTS-13 and reduces some of the adverse effects associated with fresh frozen plasma.19 Anfibatide is a potent platelet GPIb receptor antagonist derived from snake venom, which inhibits platelet aggregation.19 Additional treatment options are available or in development and are not included in this review.
Therapies for managing TTP and their mechanism of action
CONCLUSION
TTP is a rare, serious medical emergency that requires prompt diagnosis and initiation of treatment to improve patient outcomes. Prior to the 1990s, the mortality rate in TTP was greater than 90%. The addition of TPE in 1991 drastically reduced the mortality rate in TTP from greater than 90% to less than 20%. New treatment options are also available or are being investigated to improve outcomes in patients with TTP. The explosion in treatment options that are now available results from a more comprehensive understanding of the mechanism associated with TTP. This has also greatly improved diagnosis and management of the disease. The ability to measure ADAMTS-13 activity levels and the demonstration of anti–ADAMTS-13 antibodies continue to play a critical role in the diagnosis of TTP. These analytes should be readily available in laboratories with very rapid turnaround times to assist clinicians in making diagnoses. Clinicians should initiate treatment for patients who present with a high index of suspicion for TTP without waiting for confirmation from the laboratory.
Pentad of clinical symptoms associated with TTP. All 5 clinical symptoms may not be present, so thrombocytopenia and MAHA, in the absence of the remaining symptoms, are sufficient to suspect TTP and initiate therapy
1. Thrombocytopenia2. MAHA3. Fragmented erythrocytes4. Neurological deficit 5. Fever
- Received February 12, 2020.
- Revision received May 10, 2020.
- Accepted June 4, 2020.
American Society for Clinical Laboratory Science
References




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- ADAMTS-13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif - member 13
- aPTT - activated partial thromboplastin time
- BU - Bethesda unit
- EC - endothelial cell
- FDA - Food and Drug Administration
- FIB - fibrinogen
- GPIb - glycoprotein Ib
- HUS - hemolytic uremic syndrome
- IgG - immunoglobulin G
- MAHA - microangiopathic hemolytic anemia
- NAC - N-acetylcysteine
- PT - prothrombin time
- rADA - recombinant ADAMTS-13
- TMA - thrombotic microangiopathy
- TPE - therapeutic plasma exchange
- TTP - thrombotic thrombocytopenic purpura
- ULVWF - unusually large von Willebrand factor
- USS - Upshaw-Schulman syndrome
- VWF - von Willebrand factor
- VWF-CP - von Willebrand factor–cleaving protease
- WPB - Weibel-Palade body
- ADAMTS-13
- hemolytic uremic syndrome
- microangiopathic hemolytic anemia
- thrombotic microangiopathies
- thrombotic thrombocytopenic purpura
- von Willebrand factor-cleaving protease
- Upshaw-Schulman syndrome