


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Vincent Escuyer
, Wadsworth Center-New York State Department of Health, vincent.escuyer{at}health.ny.gov
LEARNING OBJECTIVES
1. Explain the current challenges with antimicrobial resistance and limitations of current testing methodologies.
2. Compare the differences between metagenomic next-generation sequencing and isolate whole-genome sequencing for antimicrobial-resistance gene detection.
3. Describe why Mycobacterium tuberculosis is an ideal candidate organism for whole-genome sequencing.
4. Discuss outcomes of Mycobacterium tuberculosis whole-genome sequencing studies and their limitations.
ABSTRACT
Increasing rates of antimicrobial resistance are a public health crisis. The emergence of resistant pathogens is multifactorial but is at least partially due to inappropriate antibiotic utilization and lack of stewardship interventions. Effective stewardship programs require timely antimicrobial resistance testing. This can be challenging for pathogens that grow slowly or not at all in culture. Next-generation sequencing approaches, such as whole-genome sequencing (WGS) of isolate, offer a more rapid alternative for such pathogens. Mycobacterium tuberculosis (TB) is a model organism for WGS to predict susceptibility due to its highly conserved and stable genome, extremely slow growth in culture, and increasing resistance rates to a limited armamentarium of anti-TB drugs. Studies have shown excellent concordance between conventional phenotypic susceptibility testing and use of WGS to predict susceptibility to at least 2 first-line anti-TB agents, rifampin and isoniazid. More data are needed for other agents, including a more comprehensive curated database of mutations paired with phenotypic data, before WGS can completely replace phenotypic testing.
- AMR - antimicrobial resistance
- CDC - Centers for Disease Control and Prevention
- DR-TB - drug resistance TB
- DST - drug susceptibility testing
- HCV - hepatitis C virus
- HIV - human immunodeficiency virus
- MDR-TB - multidrug-resistant TB
- NGS - next-generation sequencing
- PCR - polymerase chain reaction
- ReSeqTB - Relational Sequencing TB Data Platform
- TB - Mycobacterium tuberculosis
- WGS - whole-genome sequencing
- WHO - World Health Organization
- XDR-TB - extensively drug-resistant TB
INTRODUCTION
Antimicrobial resistance (AMR) is one of the major global public health challenges of this century. The emergence and spread of resistant microorganisms threaten most therapeutic and preventive options to manage bacterial infections, as commonly used antibiotics are no longer effective.1 This has a profound impact on patients, families, and, more broadly, health systems, both clinically and financially.2 Causes for the rise of antibiotic resistance include overuse in human medicine, prescribing inappropriate drugs, and extensive use in agriculture, particularly with livestock. Furthermore, the development of new antibiotics has not been prioritized by the pharmaceutical industry, mostly due to lack of profit. Therefore, very few new candidate drugs are currently in clinical trials.3 This grim situation prompted public health organizations to step in to raise awareness about the need for action to contain this global crisis. In 2013, Centers for Disease Control and Prevention (CDC) published an assessment of antibacterial threats that classifies each bacterial species in 3 distinct categories: “urgent,” “serious,” or “concerning.”4 In 2014, the World Health Organization (WHO) released a global survey on AMR surveillance from multiple national and international networks that could lay the foundation for a comprehensive plan of action.5
Managing bacterial infections is a complex process and one of the core elements is the availability of accurate methods to determine susceptibility of microorganisms to antimicrobials. However, most antimicrobial susceptibility testing is still based on culturing bacteria in the presence or absence of the antimicrobial, and turnaround time for results can be significant, particularly for slow or poor growing organisms.6 For this reason, alternative molecular methods have been or are being developed for accelerated identification and detection of resistance, either on bacterial cultures or directly on clinical specimens.7 A number of amplification-based resistance gene-detection assays are currently commercially available for many of the significant bacterial pathogens from positive blood cultures.8,9 However, most of these assays focus on the detection of very specific genetic elements or chromosomal targets and rely on correlative prediction rather than determining a true phenotype. Consequently, these rapid methods may not always produce accurate results.
The recent advent of novel sequencing technologies has circumvented this shortcoming. The emergence of several scalable and fast next-generation sequencing (NGS) platforms makes clinical diagnostic applications realistic as they can be utilized to interrogate a much higher number of targets at once, as compared with current commercially available methods.10 NGS can be performed on either DNA or RNA, allowing one to look for the presence of specific genes involved in drug resistance (DNA) or expression of these genes (RNA). Most of the current efforts focus on utilizing NGS directly on clinical specimens either with (1) multiplex polymerase chain reaction (PCR)-based amplicon sequencing with primers specifically designed to amplify a determined set of resistance genes11,12 or (2) PCR-independent shotgun metagenomics, wherein all genomic material present in the specimen is sequenced, which is followed with bioinformatics analysis to identify the resistance genes of interest.13,14 The main restriction of the former approach is the limited number of targets that are interrogated, potentially resulting in relevant genes not being included in the assay’s design. The latter approach also has several limitations. Because the respective amount of bacterial species in different specimens can greatly vary, less represented species and their resistance determinants are more likely to be undetected because of a very low amount of starting genetic material.15 Furthermore, current bioinformatics tools for metagenomics still have problems assigning resistance-associated mobile genetic elements, such as plasmids, bacteriophages, or transposons to their bacterial host genomes present in the specimen.15⇓-17 Lastly, AMR prediction in metagenomes often generates false negative results mainly due to very stringent filters, including databases mostly composed only of known, well-characterized, and clinically important resistance genes with high thresholds for sequence homology hits.15,18
The use of NGS for whole-genome sequencing (WGS) offers an attractive alternative, as it allows for interrogation of the entire genome of an organism for the presence of any resistance determinants, including cases in which more than 1 gene is involved, therefore increasing sensitivity and specificity. Prediction of AMR with WGS has already been extensively investigated mainly on clinically relevant bacteria, such as Escherichia coli, Staphylococcus aureus, Salmonella species, Streptococcus pneumoniae, Enterococcus species, Pseudomonas aeruginosa, Klebsiella pneumoniae, and Neisseria gonorrhoeae.19 A number of large, multispecies databases of resistance-determinant sequences with accompanying bioinformatics tools are publicly available and include the Comprehensive Antibiotic Resistance Database,20 ResFinder,21 or ARG-ANNOT.22 There are multiple applications of WGS for AMR, from determining the best course of treatment for infected patients to global surveillance in healthcare, community, or animal health settings to detecting foodborne dissemination of AMR.23 Most of the work published on WGS AMR thusfar relates to bacterial pathogens. However, recent studies demonstrate that this approach can also be applied to clinical virology, particularly with human immunodeficiency virus (HIV) and hepatitis C virus (HCV).24 Stanford University created an HIV drug-resistance database accessible to the general public (https://hivdb.stanford.edu), whereas University of Glasgow in the United Kingdom offers a similar service for HCV (http://hcv.glue.cvr.ac.uk/#/home). Yet, the use of WGS for clinical virology is still in development, and more work needs to be accomplished before applying it routinely in the laboratory. The remainder of this review will focus on the application to bacterial WGS for AMR, with a particular focus on Mycobacterium tuberculosis (TB).
NGS TECHNOLOGIES AND RELEVANCE TO AMR
Until the early 2000s, Sanger, or chain termination, was the main sequencing method and provided high-quality sequences, although its suitability for high throughput was not realistic for clinical application of WGS. The landscape changed dramatically with the emergence of the second and third generations of sequencing technologies, also referred to as “next-generation sequencing.”25 Second-generation sequencing is dominated by Illumina, with different instruments offering various levels of throughput. Sequencing run generates an enormous amount of nucleotide sequences, with each genome being simultaneously sequenced multiple times in small fragments (short reads) with a low per-base error rate (generally <0.1%).25 Robust bioinformatics are required for postsequencing analysis.
These short-read sequencing platforms are well suited for different applications, including amplicon-based targeted sequencing for detection of AMR determinants. However, they are not optimal for determining the sequence of closed (ie, finished) bacterial genomes, as they generate fragmented genome assemblies, also referred to as contiguous sequences or contigs, and struggle with extrachromosomal elements, such as plasmids. Sequencing of a completely closed bacterial chromosome requires the use of long-read sequencing platforms, also referred to as third-generation sequencing.26 Until recently, the market was monopolized by Pacific Biosciences with its PacBio RSII platform that comes at a high price for both instrumentation and cost per sample. In addition, this system is low throughput and is more suited for core-type facilities. Recently, Oxford Nanopore Technologies developed a long-read platform, the MinION.27 The device is slightly larger than a thumb drive and can be plugged into a standard USB port on any computer. Furthermore, sequence analysis can be performed in real-time, and results become available as soon as sequencing reads are generated during the run.28 This portability and real-time capacity have made the MinION a particularly attractive option, especially for clinical diagnostic in the field and in lower-income settings. One of the main issues with using long-read platforms for AMR is a significantly higher per-base error rate compared with short-read technology (5%–10% vs 0.1%). This rate might be too high for accurate detection of specific single-nucleotide polymorphisms associated with AMR but can partially be overcome by achieving higher read depth and improving base calling algorithms.27⇓-29 Currently, the short-read Illumina systems are the predominant platforms found in clinical laboratories.
DRUG RESISTANCE IN TB: CHALLENGES
Tuberculosis is one of the leading causes of mortality around the world. In 2017, an estimated 10 million people were newly infected with TB.30 Among them, 1.6 million died from the disease. Although the global incidence of tuberculosis is showing some decline, the emergence of drug resistance tuberculosis (DR-TB) represents a major public health crisis. Standard treatment for TB combines 4 first-line drugs: rifampin, isoniazid, pyrazinamide, and ethambutol for a period of 6 months.31 Resistance to rifampin and isoniazid, the 2 most efficient first-line drugs, results in classification of TB as multidrug resistant (MDR-TB). Treatment of MDR-TB generally involves a combination of second-line agents: fluoroquinolones and injectable medications, such as amikacin or capreomycin, in addition to other drugs chosen according to the resistance profile of targeted TB strain. Treatment can last up to 30 months. Strains that test resistant to fluoroquinolones and at least 1 injectable drug are defined as extensively drug-resistant TB (XDR-TB). Treatment of XDR cases is extremely complex, highly individualized, and often involves the use of the newest TB drugs, such as bedaquiline or delamanid, or repurposed drugs like clofazimine or linezolid.32
In 2017, WHO estimated that ~460,000 new TB cases were MDR-TB, of which ~50,000 were XDR-TB.30 In the United States, CDC classified DR-TB as a serious threat because of the complications and lower cure rates associated with long-term treatments as well as the lack of new substitute drugs to combat drug-resistant strains.4 The design and administration of an optimal drug regimen heavily rely on accurate drug susceptibility testing (DST) results.
Standard culture-based DST is impaired by the fastidious nature of TB. The very slow growth rate of the organism can significantly delay the availability of phenotypic DST results; results can take weeks to month(s) to obtain.33 Consequently, WHO proposed expanding rapid diagnosis and detection of DR-TB cases as 1 of the 5 high-priority actions to address this crisis.34 WHO endorsed 2 rapid molecular tests for detection of mutations conferring resistance to rifampin and isoniazid directly from sputum specimens: Cepheid GeneXpert MTB/RIF and Hain line probe assays.35,36 The target genes are rpoB (mutations conferring rifampin resistance) and katG and inhA (mutations conferring isoniazid resistance). Implementation of these tests in high-burden countries has partially answered the needs. However, the low number of resistance mutations targeted in these assays is a limitation, and a negative result does not necessarily indicate susceptibility to the drug of interest. Therefore, broader genetic approaches to ensure accurate diagnosis and optimal treatment for patients are needed. The ability for WGS to interrogate the entire genome rather than selected targets and derive comprehensive resistance profiles makes it an attractive alternative. It can provide physicians with important information on almost all of the currently available anti-TB drugs and guide them to optimized treatment in a timely manner, a crucial necessity with MDR-/XDR-TB infections.
TB: AN IDEAL CANDIDATE FOR WGS DST
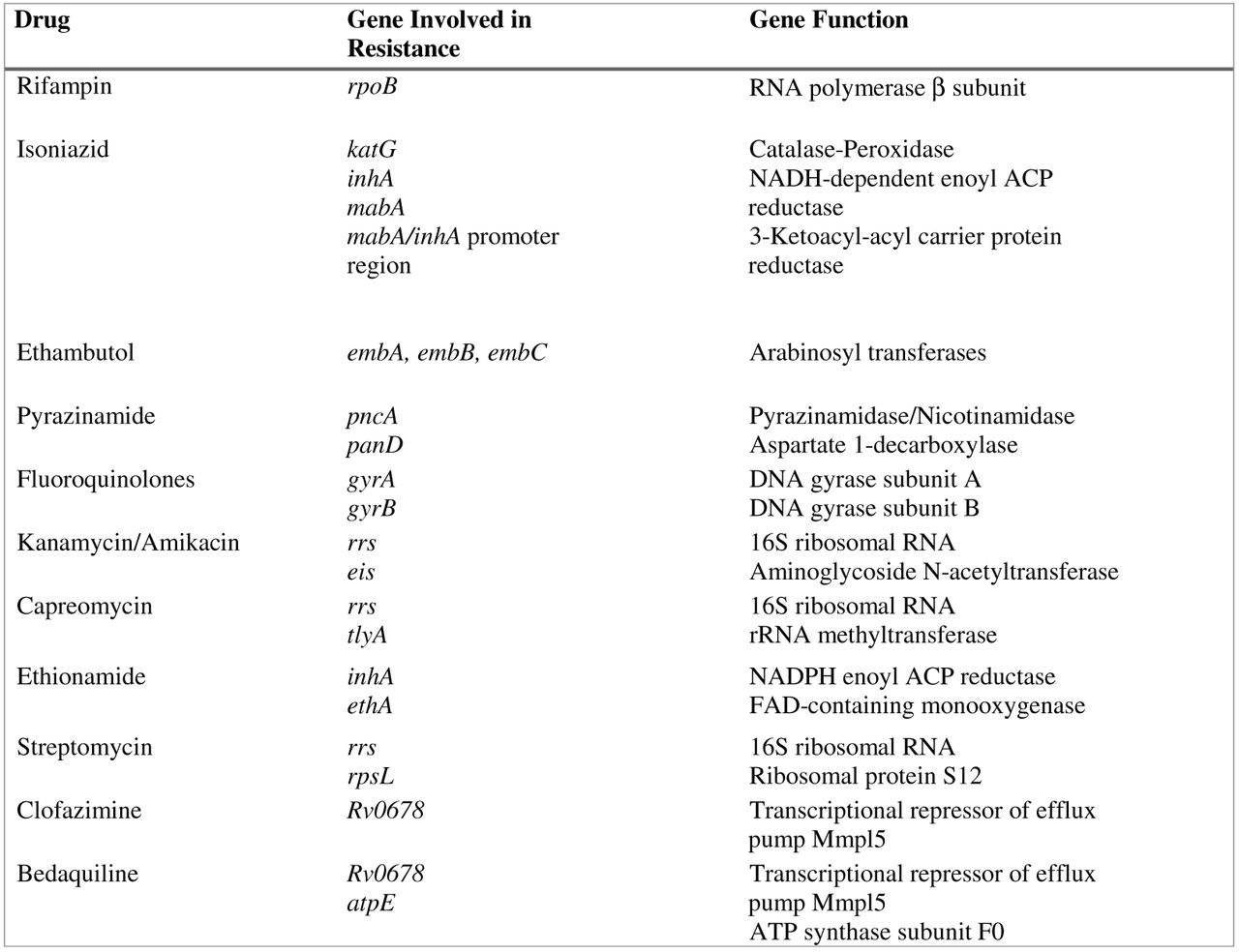
One of the main features of TB is the extreme stability of its genome with a very low mutation rate. There is no evidence of genetic material exchange between different strains nor horizontal transfer from other bacterial species. Consequently, transmissible mobile genetic elements, such as plasmids or transposons, play no role in TB drug resistance. Indeed, resistance to drugs is mostly due to the presence of mutations (single-nucleotide variations, insertions, or deletions) in specific genes that either code for the drug target itself or are involved in the activation of the drug, as is the case for pyrazinamide and isoniazid.37 Currently, most of the genes involved in drug resistance have been identified, and multiple mutations have been reported in the literature. The main genes associated with resistance are described in Figure 1. As TB treatment always involves a multidrug regimen for a long period of time, it is preferable to be able to analyze, simultaneously, multiple genes across the entire genome to generate the most comprehensive susceptibility profile, a task that no other targeted molecular approaches can achieve. Furthermore, phenotypic susceptibility testing for TB can take several weeks. For these reasons, utilization of WGS as a clinical tool for TB DST has been intensively explored the past few years and has shown great potential to improve TB clinical diagnostics by providing accurate drug-susceptibility information, timely.38⇓-40
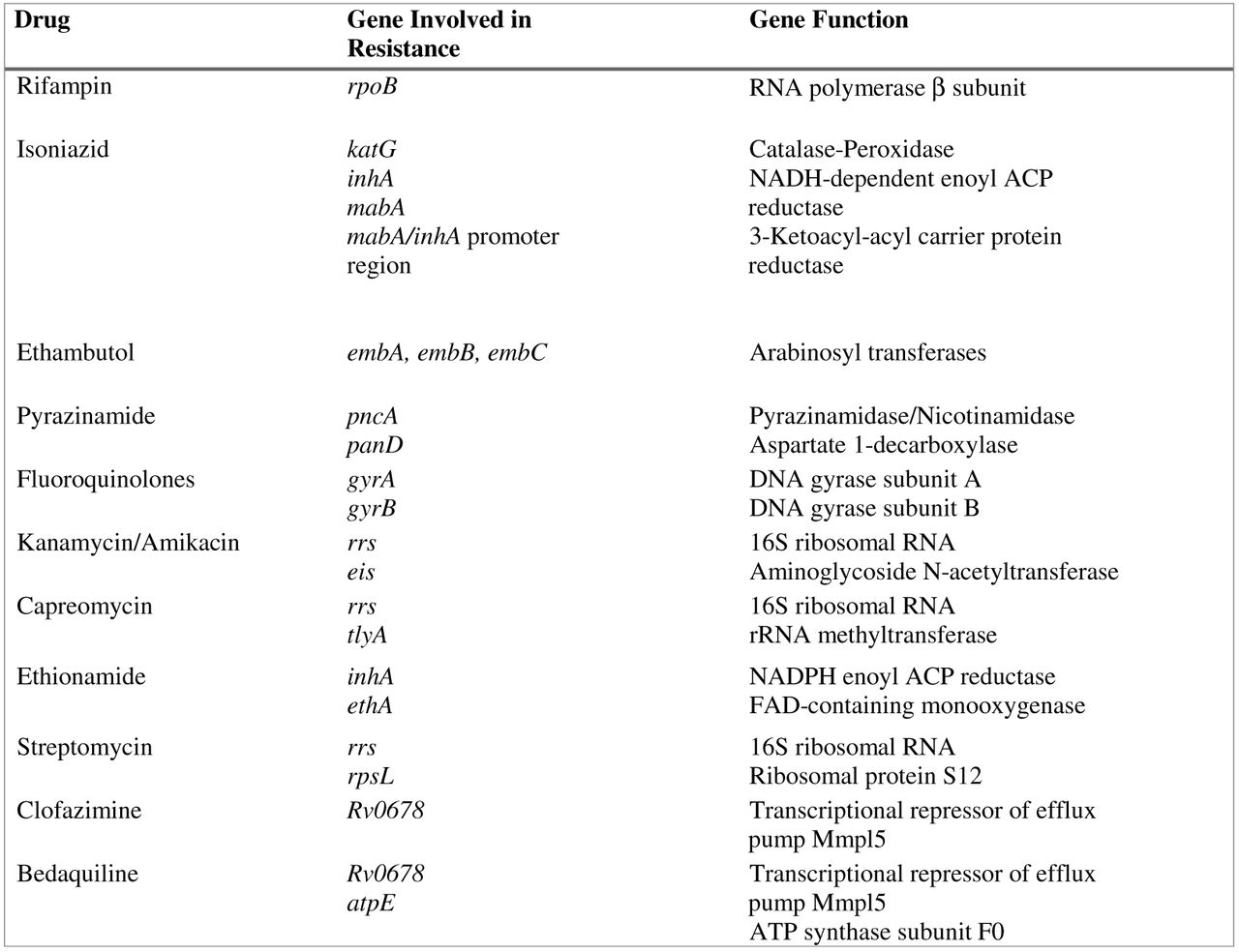
Principal genes involved in resistance to antituberculous drugs.
TB WGS WORKFLOW
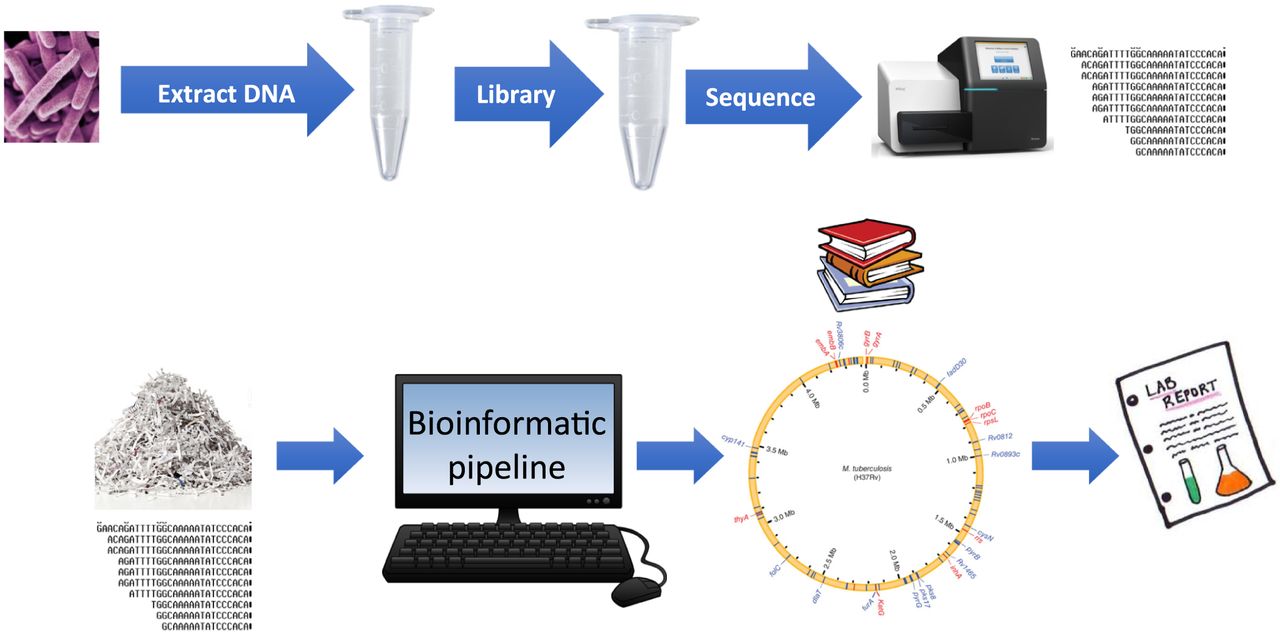
In most published studies, cultured isolates are used as the starting material. However, recent publications have explored the possibility of performing WGS directly on clinical samples to improve turnaround time. Workflow for TB WGS is summarized in Figure 2 and always includes similar steps independent of the sequencing platform utilized. DNA extraction is a crucial step, as the quality and quantity of the extracted DNA will determine the quality of the sequencing data.41 Several lysis methods can be used and include chemical, mechanical, thermal, or any combination of the above. Subsequently, a DNA library is prepared in which genomic DNA is fragmented and specialized adapters are attached at both ends of the DNA fragments. Numerous kits to prepare sequencing libraries are available commercially from different vendors. Adapters are specific for the sequencing platform utilized, and adapter ligated fragments are then amplified by PCR, purified, and loaded into the sequencer.42 At the end of the sequencing run, an enormous amount of sequence data is generated that requires significant computational resources for analysis and storage.43 This requirement is often a major barrier for clinical laboratories interested in adding this technology in their workflow. As with the other reviews in this Focus series, there are currently no Food and Drug Administration–approved approaches to WGS for AMR in TB or any other pathogen. Thus, there is a large burden on the adopting laboratory to undertake an expensive, complex, and time-consuming validation.

General workflow for TB WGS.
BIOINFORMATICS TOOLS
Prediction of resistance by WGS requires large databases of mutations associated with drug resistance and software able to interrogate these databases for the presence of these mutations to generate a drug-resistance profile. A number of online tools publicly available have been developed during the past decade. The first extensive database was the Tuberculosis Drug Resistance Mutation Database.44 However, this database has not been updated since April 2010 but has provided the foundation for the construction of newer databases. Since 2017, The Rapid Drug Susceptibility Testing Consortium, part of the Critical Path to TB Drug Regimens Initiative, opened their Relational Sequencing TB Data Platform (ReSeqTB) to public.45 This tool has a user-friendly interface to access WGS data collected from multiple private and public databases combined with culture-based drug susceptibility information and clinical outcome data when available.46 Currently, the main web-based tools specifically designed for TB sequence analysis include PhyResSE, an integral component of ReseqTB platform; genTB; and TBProfiler.47⇓-49 In addition, 2 software solutions are available as well, Mykrobe Predictor TB and MTBSeq.50,51 Criteria for selecting a particular bioinformatics platform can vary among clinical laboratories depending on their respective regulatory requirements for clinical validation.
PERFORMANCES OF TB WGS
Multiple studies using WGS to determine resistance profiles with TB have been published, with thousands of TB strains sequenced to date. Data from these studies showed excellent concordance with culture-based phenotypic drug-susceptibility results.52⇓⇓-55 A large study published by the CryPTIC Consortium on more than 10,000 isolates demonstrates that this is particularly true for the 2 main first-line drugs, rifampin and isoniazid, for which an overall specificity of 0.94 and a sensitivity of 0.94 and 0.93, respectively, were found.56 More variations were observed with the other first-line drugs ethambutol and pyrazinamide, with specificities of 0.84 and 0.92 and sensitivities of 0.86 and 0.76, respectively. An incomplete understanding of the mechanisms underlying resistance to these drugs could partly explain this performance.37,57 Performance data for drugs other than the first-line drugs, which are only used in case of demonstrated resistance to the standard drugs, are sparse, although WGS performs well with fluoroquinolones.58 This lack of information could be explained in part by the paucity of phenotypic data for these drugs, as their testing is mostly performed in specialized laboratories and is not broadly available.
Most of the studies published thusfar were retrospective, so more prospective work is needed to fully evaluate the performance of WGS in a clinical setting. A few prospective studies have been reported and showed promising results.59⇓⇓-62 For example, in New York, Shea et al tested prospectively 405 clinical isolates over 1 year and found susceptibility and sensitivity values similar to those reported in the retrospective studies, strongly suggesting the possibility to implement WGS as a replacement for conventional DST. Prospective data from these studies indicated a particularly high concordance between WGS prediction and susceptibility to TB drugs. Consequently, both Public Health England and New York State Department of Health recently decided to move forward and use WGS as the primary method to replace phenotypic susceptibility testing for all clinical isolates predicted to be fully susceptible by WGS. However, a similar change for isolates predicted to be resistant by WGS is not yet ready for implementation. It has been shown that some mutations detected in target genes do not confer phenotypic resistance, which could cause an isolate to be improperly reported as resistant to a particular drug. Consequently, a physician might alter treatment by eliminating an important drug that would have still been appropriate per conventional testing. This emphasizes the need for a better understanding of resistance mechanisms in TB as well as carefully curated mutation databases that are not yet available.
REMAINING CHALLENGES
A number of key issues, both technical and logistical, remain before being able to generalize the implementation of WGS in routine clinical testing. This starts with the necessity of standardizing procedures for sample preparation, including methodology for DNA extraction, quantification, and quality control. One of the current limitations of WGS is still the need for culture before sequencing. Consequently, turnaround time for susceptibility results is impacted by this requirement due to the fastidious nature of the organism that can take days to weeks to grow in culture. Recent studies have started to address this issue and explored the feasibility of using WGS directly on clinical specimen to predict drug resistance, thereby bypassing culture.63⇓⇓-66 However, these approaches require users to enrich TB DNA from the specimen to a level of concentration and purity suitable for WGS and involve often-costly systems and labor-intensive protocols that are impractical for routine clinical testing.
Although WGS has been shown to perform very well to predict resistance to first-line drugs and fluoroquinolones, there is still a lack of data for the second-line drugs and all new or repurposed drugs used to treat MDR-/XDR-TB, such as linezolid, clofazimine, or bedaquiline. Better understanding of the mechanisms of resistance to these drugs is still a work in progress, and WGS performance will improve as new mutations are identified.67,68 However, as bioinformatics pipelines progress by integrating new information, clinical laboratories will be required to revalidate these pipelines as they are updated, a process that could be cumbersome.
CONCLUSION
There is a crucial necessity to advance the methods of identification of AMR and DST for TB and other microbial pathogens in the clinical setting. WGS represents an attractive alternative to other conventional and molecular methods. It has the potential to transform TB DST in the clinical laboratory, thus having a profound impact on patient management and public health intervention by providing comprehensive results significantly faster than culture-based methods. Significant progress has already been made on this front, and many tools are now available to the clinical laboratories and physicians. The NGS industry is starting to evolve toward developing ease-of-use and lower-cost technology, but affordability and complexity remain as obstacles for implementation in low- to middle-income countries. In this regard, the MinIOn sequencing platform could address some of these issues by offering an affordable and portable “easy to use” device. Similar ease-of-use advances would be needed for specimen and library preparation, and bioinformatics for data analysis before developing countries could realistically adopt such testing. Nevertheless, as knowledge and technology are making strides in reaching these goals, it is reasonable to anticipate that within the next decade, industrialized countries will replace conventional TB DST with WGS. Change will be more gradual in developing countries where WGS will mostly be used more sporadically as a support methodology for drug-resistance surveillance and development of new rapid diagnostics.
- Received June 1, 2019.
- Accepted September 5, 2019.
American Society for Clinical Laboratory Science
References




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- AMR - antimicrobial resistance
- CDC - Centers for Disease Control and Prevention
- DR-TB - drug resistance TB
- DST - drug susceptibility testing
- HCV - hepatitis C virus
- HIV - human immunodeficiency virus
- MDR-TB - multidrug-resistant TB
- NGS - next-generation sequencing
- PCR - polymerase chain reaction
- ReSeqTB - Relational Sequencing TB Data Platform
- TB - Mycobacterium tuberculosis
- WGS - whole-genome sequencing
- WHO - World Health Organization
- XDR-TB - extensively drug-resistant TB
- whole-genome sequencing
- next-generation sequencing
- antimicrobial resistance
- Mycobacterium tuberculosis