


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Larry J. Smith
, Division of Hematology, Abbott Diagnostics, larry.smith{at}abbott.com
LEARNING OBJECTIVES
1. Compare and contrast the differences between hemophila A and B.
2. Explain the pathogenesis and genetic mechanisms associated with the disease.
3. Describe clinical and laboratory manifestations observed in patients.
4. List treatment options for treating patients with hemophilia.
ABSTRACT
Hemophilia is a rare, congenital bleeding disease with an X-linked recessive inheritance pattern. It is characterized by absent, decreased, or dysfunctional coagulation factor VIII (FVIII) or factor IX (FIX). Individuals with severe hemophilia bleed into the joints, soft tissue, and muscles, which can be debilitating. Although hemophilia is a serious life-long bleeding disease, understanding the pathogenesis of the disease leads to better patient treatment and improved patient outcomes.
- AHA - acquired hemophilia A
- aPTT - activated partial thromboplastin time
- FIIa - thrombin
- FIX - factor IX
- FV - factor V
- FVIII - factor VIII
- FX - factor X
- FXI - factor XI
- HA - hemophilia A
- HB - hemophilia B
- HTC - hemophilia treatment center
- PT - prothrombin time
- TF - tissue factor
- TFPI - tissue factor pathway inhibitor
- VWF - von Willebrand factor
- acquired bleeding disorders
- acquired factor VIII deficiency
- bleeding diathesis
- Christmas disease
- bleeding
- congenital bleeding disorders
- hemophilia
- hemophilia A
- hemophilia B
INTRODUCTION
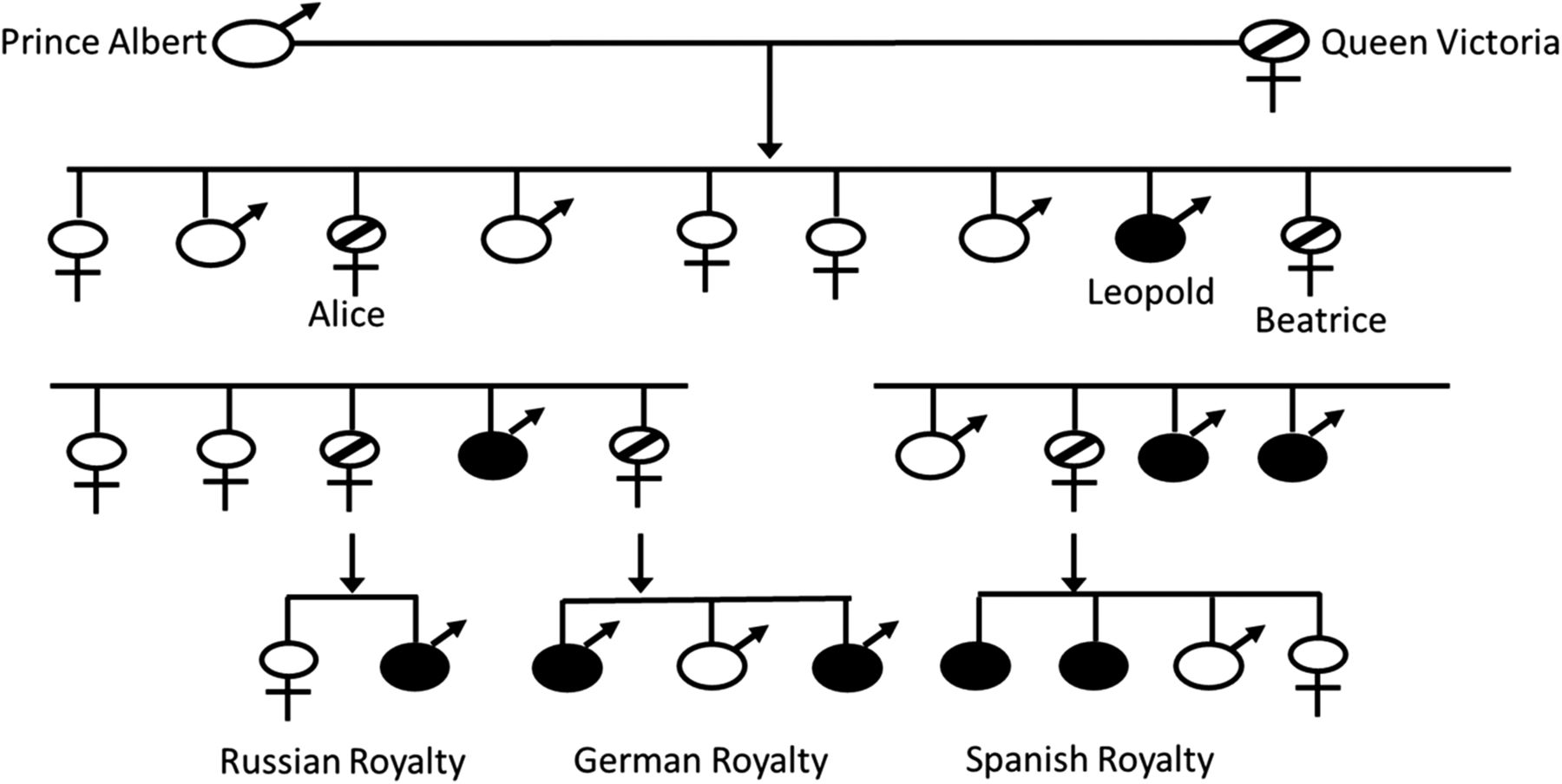
Hemophilia is a recessive, X-linked bleeding disease that results from a decreased or absent level of coagulation factor VIII (FVIII; hemophilia A), a decreased or absent level of coagulation factor IX (FIX; hemophilia B), or to the inability for either of these coagulation factors to participate in the coagulation cascade. The disease can be congenital or acquired. The prevalence of congenital hemophilia A (HA) is 1 in 5,000 live male births and for hemophilia B (HB) it is 1 in 30,000 live male births.1 The true prevalence may actually be a little higher owing to milder forms of the disease not being diagnosed in many parts of the world. Initial descriptions of hemophilia date back to as early as the second century in the Talmud, which state that male babies should not be circumcised if 2 previous male siblings died of excessive bleeding as a result of circumcision.2 Albucasis later described a family in which males died following minor injuries, and John Conrad Otto is credited with the first modern description of the disease in 1803.2,3 Hemophilia has also been referred to as the “royal disease” because various members of the royal families were affected by hemophila. Queen Victoria of England is reported to have been a HB carrier. She had 1 son—Leopold—who had hemophilia and 2 daughters—Alice and Beatrice—who were carriers and transmitted the disease to the Russian, German, and Spanish royal families (Figure 1).1,4 The concept of 2 different hemophilias was suggested by Pavlovsky5 when he demonstrated that blood from 1 patient with hemophilia could correct the clotting problem in a second patient with hemophilia and vice versa.

The “Royals” family tree. Queen Victoria is reported to have been a carrier of HB. Alice and Beatrice transmitted the disease to the Russian, German, and Spanish royal families.
HA and HB are clinically indistinguishable from each other, but they differ in the genetic mutation that leads to the bleeding disease. They also differ in the frequency of inhibitors that develop, quality of life, and patient treatment. Proper diagnosis can be made by performing coagulation factor assays for FVIII and FIX in the clinical laboratory. In both diseases, the activated partial thromboplastin time (aPTT) is prolonged, while the prothrombin time (PT) is normal; however, the activity level of FVIII is decreased/absent in HA and the activity level of FIX is decreased/absent in HB. The hemophilias are further classified as mild (0.05–0.40 IU/mL, [>5%–<40%]), moderate (0.01–0.05 IU/mL, [1%–5%]), and severe (<0.01 IU/mL, [<1%]). The bleeding phenotype generally corresponds to the severity of the factor level (Table 1).6 Patients with severe hemophilia develop spontaneous and recurrent bleeds without obvious injury or trauma, while patients with moderate and mild hemophilia bleed after injury, trauma, or surgical procedures. The hallmarks of bleeding seen in patients with hemophilia include hemarthrosis (bleeding into the joints) and musculoskeletal bleeding, which involves the muscles and soft tissue. According to the National Hemophilia Foundation, the percentage breakdown of overall hemophilia population by severity is 60%, 15%, and 25% for severe, moderate, and mild deficiencies.7
Clinical bleeding versus factor activity
PATHOPHYSIOLOGY OF HEMOPHILIA
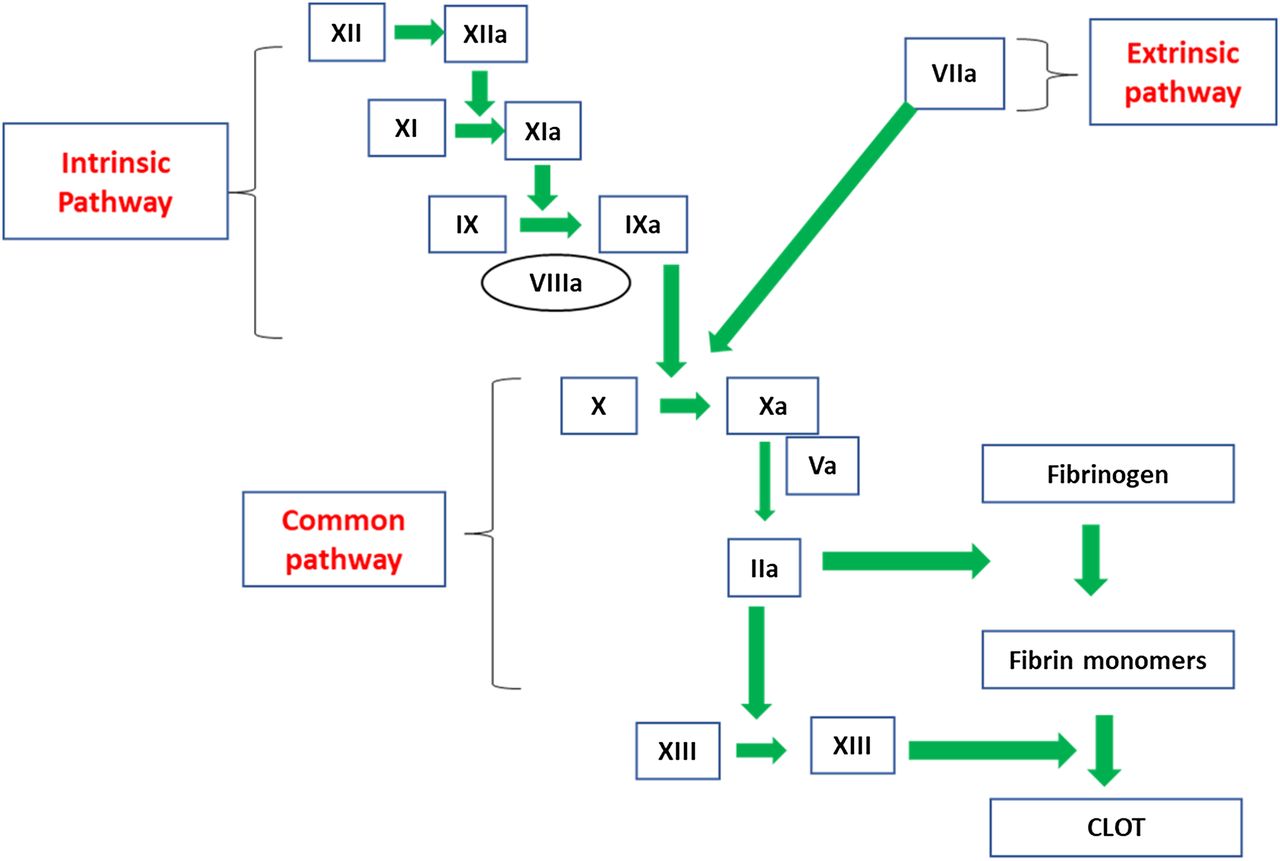
FVIII and FIX are located in the intrinsic pathway of the coagulation cascade and play a role in the formation of the intrinsic-tenase complex upon activation (Figure 2). FVIII is known as the antihemophilic factor, and FIX is known as the plasma thromboplastin component or Christmas factor. Immediately following tissue damage, tissue factor (TF) is released into the lumen of the blood vessel and binds to FVII in the circulation to form a TF-VIIa complex. This complex activates factor X (FX), converting it to FXa, and activates FIX to FIXa.8 FIXa, along with FVIIIa, convert FX to FXa. FVIIIa serves as a cofactor in this reaction, increasing the rate of conversion of FX to FXa by FIXa.9 Activated FX, along with FVa, converts prothrombin to thrombin (FIIa), and FIIa converts fibrinogen to fibrin to form an initial clot. In addition to contributing to fibrin clot formation, FIIa activates factor XI (FXI) to FXIa, FVIII to FVIIIa, and factor V (FV) to FVa. After FXIa converts FIX to FIXa, the increased levels of FIXa are available to interact with the additional FVIIIa, which leads to continued FIIa generation via the intrinsic pathway. The presence of FVIIIa and FIXa are essential for this continued FIIa generation because the extrinsic pathway is downregulated very early during the activation phase of the coagulation cascade. The participation of both extrinsic and intrinsic pathways is needed to form a solid, stable, and protective clot. When either FVIII or FIX are absent, severely decreased, or defective, the clot that forms is insufficient to support normal hemostasis.10 Sixma et al11 performed histological examinations of clots from patients with hemophilia and demonstrated that the outer region of the clots from hemophiliac patients were stabilized by a fibrin meshwork; whereas, the inner portion of the clot showed little or no fibrin formation. Therefore, hemophilia results from an inability to prolong FIIa generation via the intrinsic pathway owing to absent, decreased, or abnormal production of either FVIII or FIX.
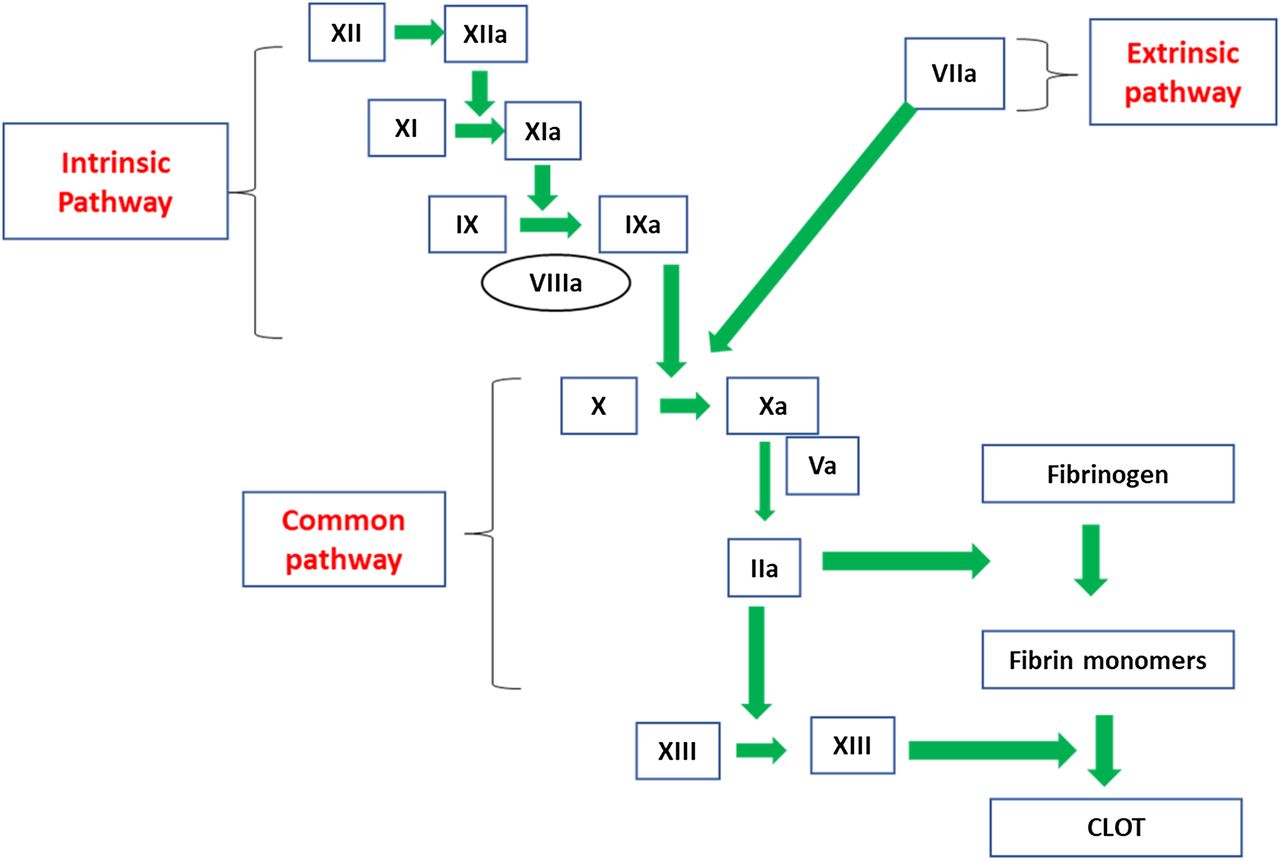
The coagulation cascade. FVIII and factor VIX are located in the intrinsic pathway.
The cell-based model of coagulation adds further insight into the pathogenesis of hemophilia. This model includes platelet and cell surfaces that are essential in in vivo coagulation. The cell-based model of coagulation consists of 3 phases to describe coagulation: (1) initiation phase, (2) amplification phase, and (3) propagation phase. During initiation, TF-bearing cells expose TF to FVII in the plasma, creating the TF:FVIIa complex that activates FX and FIX. FIXa migrates to the platelet surface while FX remains on the surface of the TF-bearing cell.12 FXa on the tissue-bearing cell is inhibited by tissue factor pathway inhibitor (TFPI). Amplification occurs on the platelet surface. During amplification, small amounts of FIIa generate activated FV, FVIII, and FXI. The propagation phase follows whereby the intrinsic-tenase complex (FVIIIa/FIXa) and the prothrombinase complex (FXa/FVa) form on the platelet surface. FXa on the platelet surface is protected from inhibition by TFPI. A burst of FIIa is generated that converts fibrinogen to fibrin and activates FXIII, which stabilizes the fibrin clot. Formation of both the tenase and prothrombinase complex is critical for prolonged FIIa generation. In patients with hemophilia, an absent/defective FVIII or FIX results in a nonfunctional intrinsic-tenase complex on the platelet surface. Although FXa is generated in patients with hemophilia, it is not able to move from the TF-bearing cell to the activated platelet surface to contribute to FIIa generation.12,13
GENETICS OF HEMOPHILIA
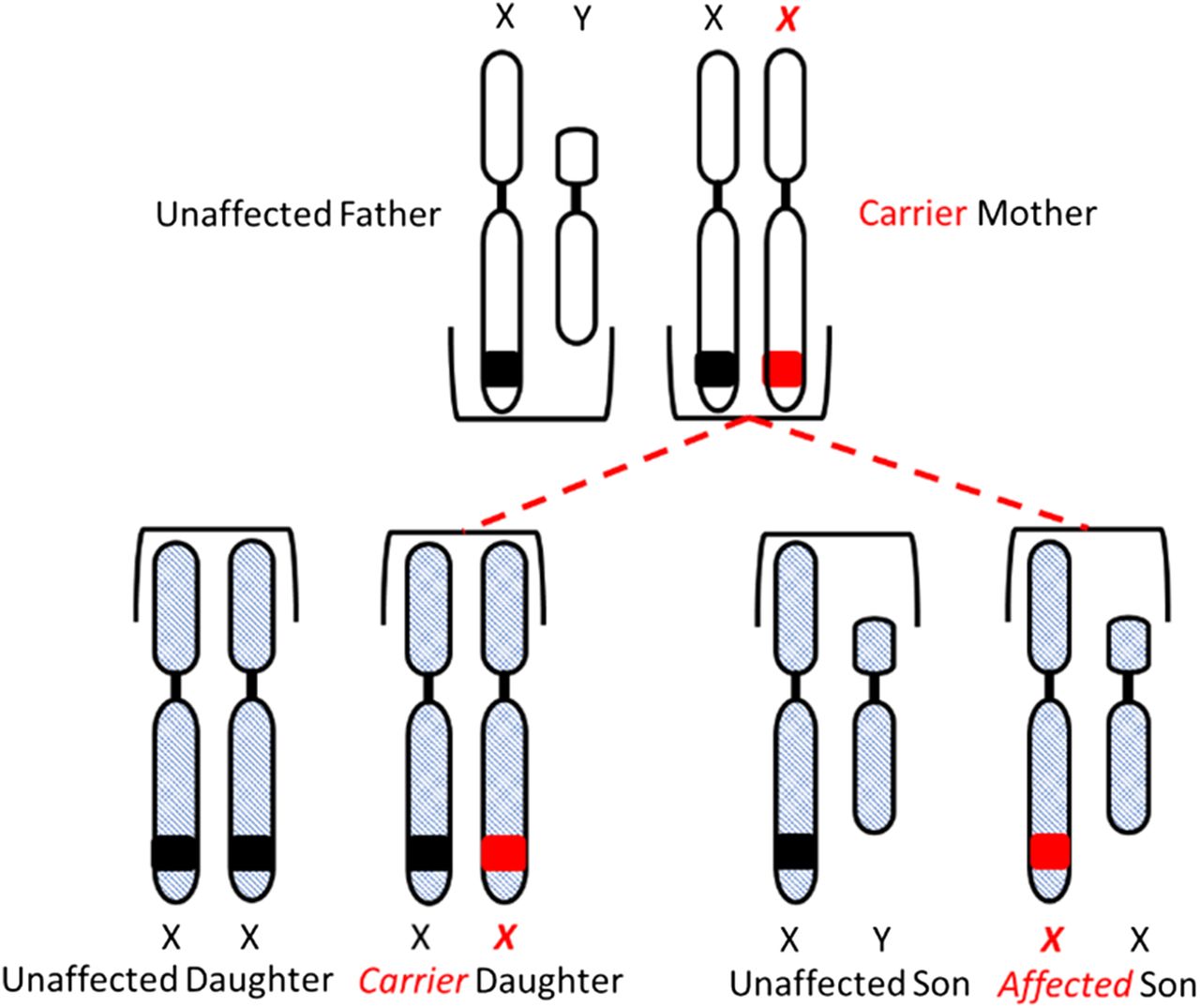
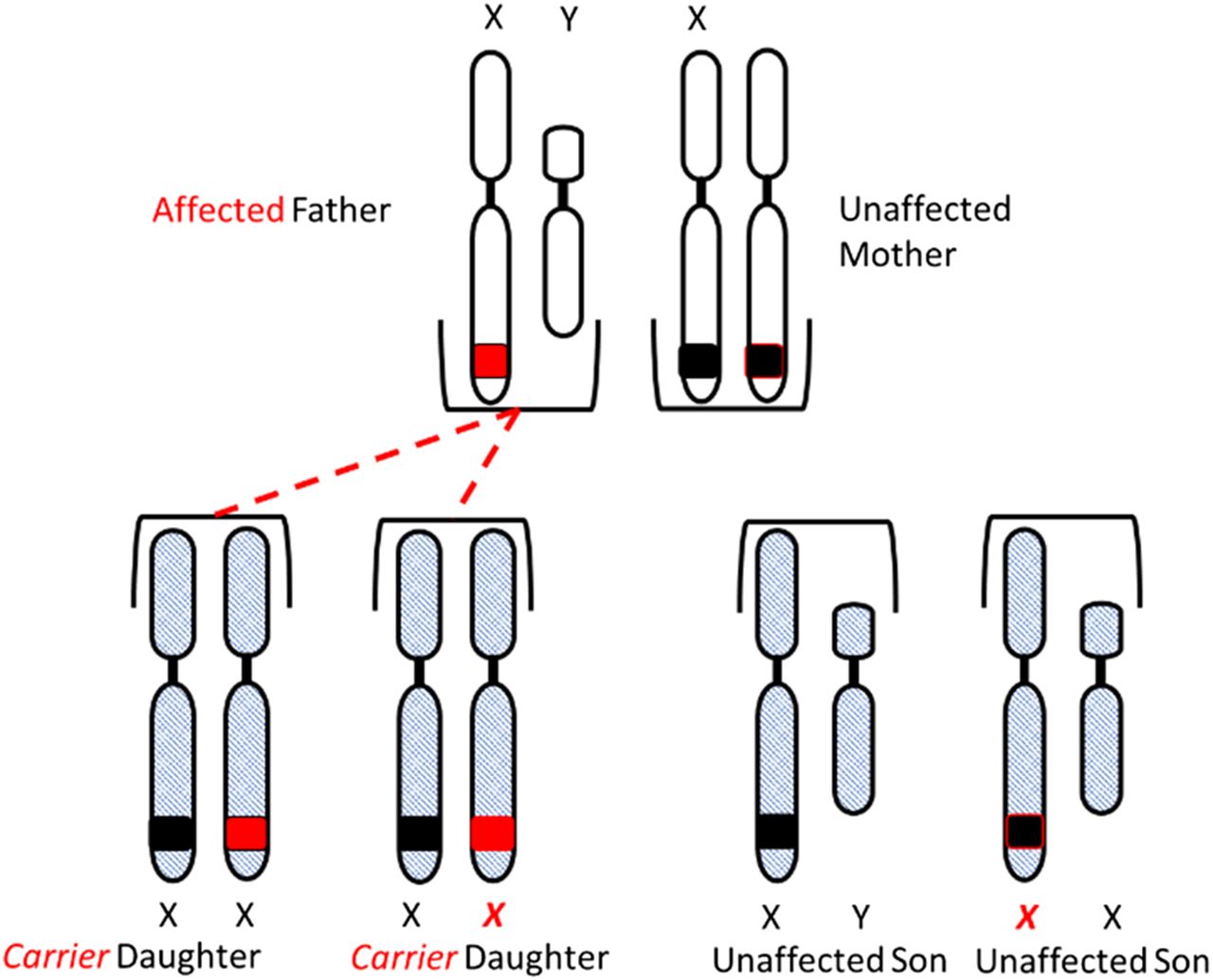
Congenital HA and HB are X-linked diseases inherited in a recessive pattern. Sons born to a mother who is a carrier of the gene and an unaffected father have a 50% chance of inheriting the disease, and daughters have a 50% chance of becoming carriers of the gene. All daughters born to an unaffected mother and a hemophiliac father will be carriers of the gene, and none of the sons will be affected (Figure 3A and 3B).
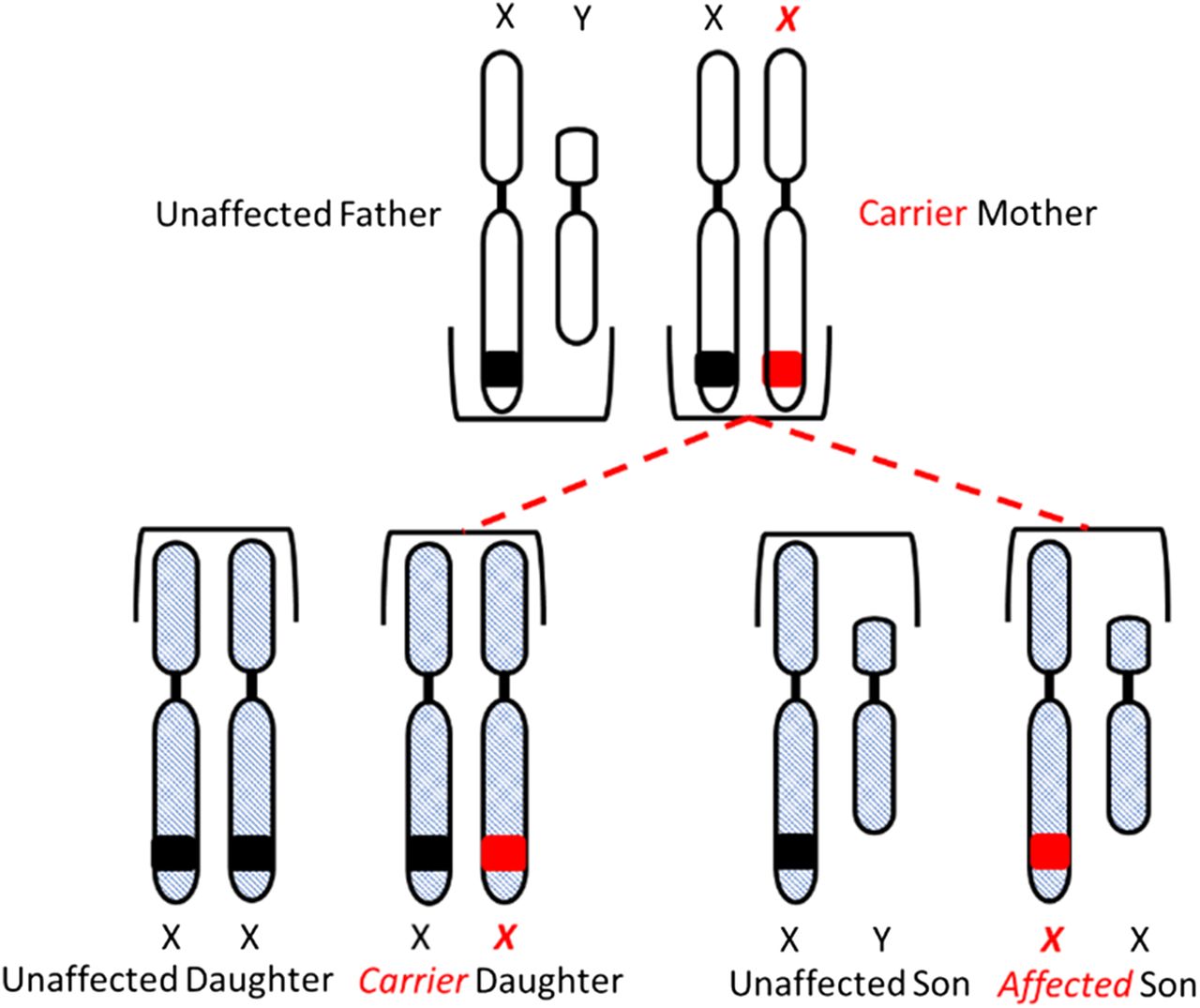
Inheritance of hemophilia. Daughters have a 50% chance of inheriting the gene from a carrier mother and unaffected father.
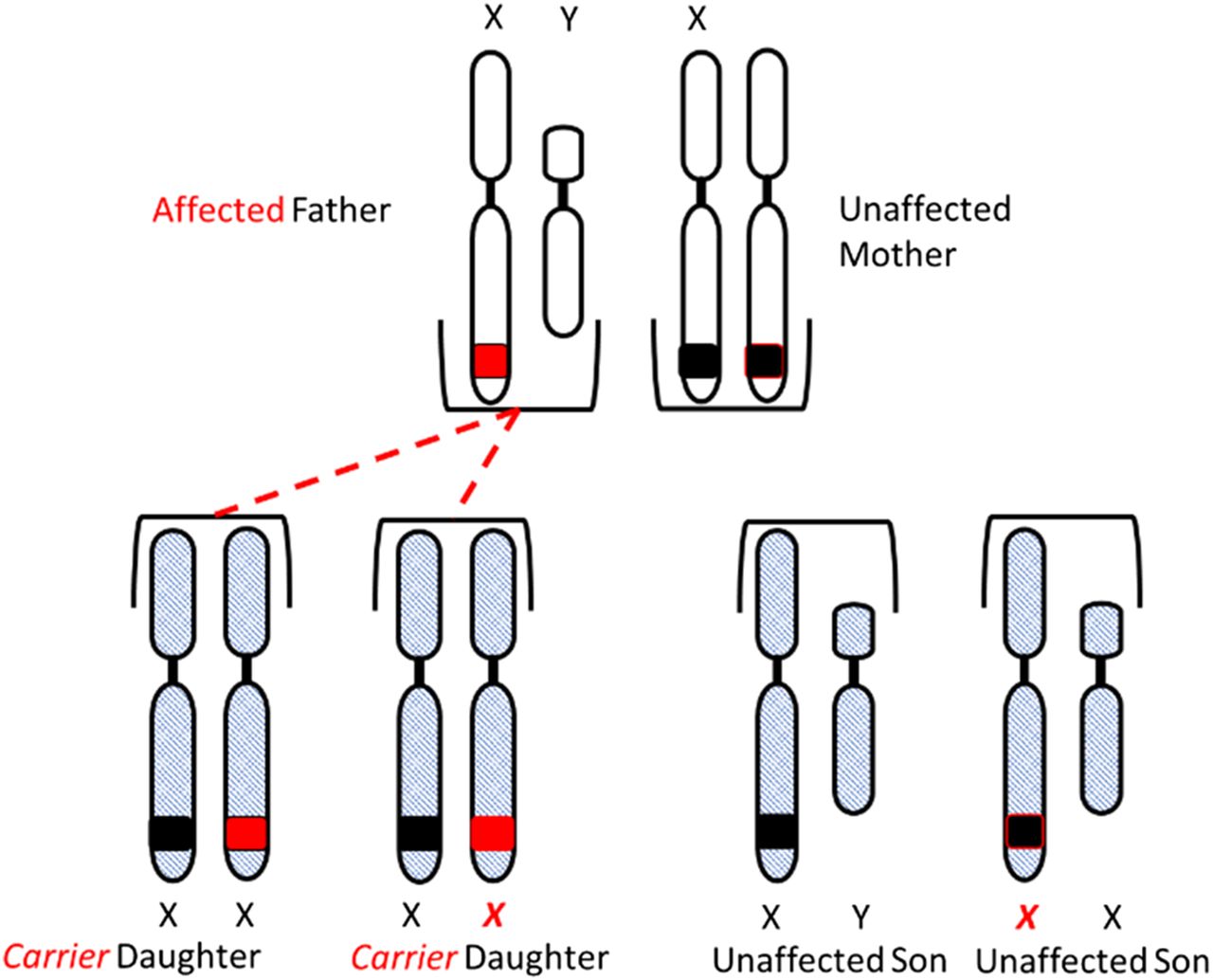
Inheritance of hemophilia. All daughters of an affected father and unaffected mother will inherit the gene.
Additionally, up to 30% of cases may arise de novo, resulting from a spontaneous mutation in the absence of a family history of hemophilia.1 There are reports, however, that in up to 80% of the de novo mutations, genetic testing of the mothers demonstrated that they were carriers of the gene.14 The genes encoding FVIII and FIX are located on the tip of the long arm of the X chromosome: Xq28 and Xq27, respectively.14,15
The genes for FVIII and FIX were cloned in 1984 and 1982, respectively.16 The FVIII gene is 186 kb and contains 26 exons that code for a signal peptide and a 2332-amino acid polypeptide. The polypeptide contains 6 domains (A1-A2-B-A3-C1-C2).17 FVIII synthesis occurs in liver sinusoidal-endothelial cells and vascular-endothelial cells. FVIII circulates as a heterodimer bound to von Willebrand factor (VWF). Upon activation by FIIa, FVIII is released from VWF and interacts with FIXa and FXa on the phospholipid surface of activated platelets.14 The gene for FIX is 33.5 kb and contains 8 exons that code for a 415-amino acid polypeptide.17 FIX is synthesized in liver hepatocytes.14 FIX is a vitamin K-dependent protein that requires γ-carboxylation before becoming functional. More than 2,000 unique molecular defects have been described in the FVIII gene, and about 1,095 genetic variants have been described in the FIX gene.6,18,19 The range of mutations for HA and HB include point mutations, insertions, deletions, and inversions. Inversion of intron 22 on the X chromosome is the most common mutation observed in severe HA and accounts for about 45% of cases. The inversion is spontaneous and occurs during meiosis in male germ cells. A similar type of inversion occurs in intron 1 and accounts for 2%–5% of cases of severe HA.20 It has been suggested that the reduced frequency of HB compared to HA may be because of the smaller size of the genes involved: 33.5 kb for FIX and 187 kb for FVIII.
ACQUIRED HEMOPHILIA
Acquired hemophilia A (AHA) is a rare autoimmune disease and affects approximately 1.5 million individuals annually. It is associated with a high risk for morbidity and mortality.21 It is predominantly seen in elderly individuals and equally distributed between males and females. It is also associated with pregnancy and individuals with autoimmune disease. AHA occurs when spontaneous production of neutralizing IgG antibodies target FVIII, which prevents it from functioning in the intrinsic-tenase complex.22 The autoantibodies seen in AHA are different from the alloantibodies that result from exogenous FVIII therapy in patients with congenital HA.23
Bleeding episodes in patients with AHA often are more severe than in patients with congenital HA, in part because of the kinetics of inactivation of FVIII autoantibodies. Alloantibodies typically inactivate FVIII in a linear fashion (type I, or first-order kinetics) and are concentration- and time-dependent. In addition, alloantibodies completely inactivate FVIII. Autoantibodies, on the other hand, have a rapid inactivation phase followed by a slower equilibrium phase (type II or second-order kinetics).23 Autoantibodies incompletely inactivate FVIII. Patients with AHA usually do not have a history of bleeding like patients with congenital hemophilia; therefore, early recognition and intervention is essential.
CLINICAL AND LABORATORY MANIFESTATIONS
Clinical symptoms seen in patients with HA or HB are identical with bleeding as the hallmark. The severity and type of bleeding varies and depends on the severity of the coagulation factor deficiency (Table 1). Bleeding tendencies may also vary with the age of the patient. Neonates with hemophilia may experience intracranial hemorrhage at a rate of 40–80 times higher than the average population.14 Circumcision may also pose a significant risk of bleeding in cases of severe hemophilia. In children and adults with severe hemophilia, the incidence of musculoskeletal bleeds increases as these individuals are more active and are prone to incidental trauma. These musculoskeletal bleeds contribute to the long-term bleeding complications seen in individuals with severe hemophilia.14 One way to minimize bleeding in individuals with severe hemophilia is early initiatation of prophylaxis. Prophylaxis involves administration of factor concentrate on a regular basis to maintain adequate factor levels to prevent bleeding. Individuals with moderate to mild hemophilia experience fewer bleeding events and require more significant trauma to initiate bleeding. The amount of bleeding seen in moderate to mild hemophilia correlates with the level of factor activity.
Hemarthrosis is very common and accounts for about 75% of bleeding events in hemophilia.14 Hemarthrosis tends to repeat in the same joint. The joints most commonly affected are the ankles, knees, and elbows. The release of hemoglobin from the erythrocytes ultimately triggers an inflammatory event, leading to synovitis. As these events occur on multiple occasions in the same joint, the synovium undergoes thickening, and the bone and cartilage become destroyed, which leads to arthropathy.24 Musculoskeletal bleeding and mucosal bleeding (including epistaxis) are commonly seen as well.25
The development of inhibitors is a serious iatrogenic complication associated with hemophilia treatment. Inhibitors are specific antibodies (IgG) that develop against coagulation FVIII or FIX preparations used in therapy. These antibodies are neutralizing antibodies that inhibit the procoagulant activities of FVIII and FIX. The development of inhibitors is more commonly seen in HA (25%–30% of patients) compared to HB (3%–5% of patients).24 The lower frequency of inhibitors may in part be because of increased immunogenicity of FVIII compared to FIX.
A prolonged aPTT with a normal PT is the classic finding in the clinical laboratory among patients with hemophilia. The prolongation of the aPTT is directly related to the factor activity levels of FVIII and FIX in the patient. In addition to a prolonged aPTT, laboratories often refer to a mixing study as part of the algorithm for working up a prolonged aPTT. If an inhibitor has been identified, a laboratory will perform a Bethesda assay to quantitate the antibody titer in the patient sample. A more thorough review of the role of the laboratory in working up hemophilia can be found in the accompanying article, “Laboratory Monitoring for Hemophilia.”
PATIENT TREATMENT
Patients who present with bleeding should be treated promptly to avoid severe complications. In addition to being treated for bleeding, these patients should be treated for additional health and psychosocial needs. A hemophilia treatment center (HTC) offers a multidisciplinary approach that takes into consideration various life stages of the patient. The multidisciplinary team includes hematologists, orthopedists, laboratory scientists, nurses, physical therapists, social workers, and dentists. Treatment and management protocols are individualized based on the patient’s age, weight, bleeding pattern, joint health, physical activity, clotting factor levels, and compliance.26 It has been reported that patients using an HTC are 40% less likely to die of a hemophilia-related complications and are less likely to be hospitalized for bleeding complications.27⇓-29 Treatment of bleeding problems may consist of hemostatic support, adjunctive therapies, prophylaxis, and/or inhibitor management. Hemostatic support involves the use of exogenous-hemostatic agents to increase the level of deficient FVIII or FIX to an adequate level in the patient to prevent further bleeding. A more in-depth review of the therapeutic agents used to treat patients with hemophilia are found in the accompanying article, “Past, Present and Future Options in the Treatment of Hemophilia A,” in this focus series.
CONCLUSION
Hemophilia is a very complex bleeding disease that once was once considered a debilitating disease. During the first half of the twentieth century, the life expectancy of patients with hemophilia ranged from about 15 to about 24 years. Today, these patients lead near typical lives because of proper medical care, education about their disease, and the combined efforts of a quality health care team. Great strides have been made in the development of novel hemostatic drugs with extended half-lives that reduce the need for more frequent administration. These drugs have also been optimized for improved safety and efficacy. This has led to measurable reductions in the major complications associated with severe hemophilia. However, the development of inhibitors remains an issue in treating patients with severe hemophilia. The risk of infectious diseases (human immunodeficiency, hepatitis B, and hepatitis C) transmitted through the use of blood products was very high in the early 80s but has been eliminated through improved techniques for handling and screening of blood products and the advent of recombinant replacement products. While these products improve patient adherence and quality of life, they do not provide a “cure.” The only potential cure for hemophilia is gene therapy. However, the challenge remains to develop a safe, viable gene therapy product that is not cost-prohibitive. There are additional therapeutic agents on the horizon that down-regulate the naturally-occurring inhibitors of coagulation, leading to improved hemostasis. These agents include, but are not limited to, (1) molecules that inhibit TFPI; (2) molecules targeting antithrombin; and (3) a bispecific antibody that targets FIXa and FX, which mimicks the action of FVIIIa. The progress that has been made to understand and treat hemophilia is tremendous, but it does not come without cost. The goal will be to continue improvements in safety and efficacy, reduce cost, and increase availability for all individuals worldwide.
- Received October 18, 2018.
- Accepted March 13, 2019.
American Society for Clinical Laboratory Science
REFERENCES




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- AHA - acquired hemophilia A
- aPTT - activated partial thromboplastin time
- FIIa - thrombin
- FIX - factor IX
- FV - factor V
- FVIII - factor VIII
- FX - factor X
- FXI - factor XI
- HA - hemophilia A
- HB - hemophilia B
- HTC - hemophilia treatment center
- PT - prothrombin time
- TF - tissue factor
- TFPI - tissue factor pathway inhibitor
- VWF - von Willebrand factor
- acquired bleeding disorders
- acquired factor VIII deficiency
- bleeding diathesis
- Christmas disease
- bleeding
- congenital bleeding disorders
- hemophilia
- hemophilia A
- hemophilia B