


Article Figures & Data
Figures
- Figure 1.
Conventional and NGS methods for viral outbreak investigation. The Sanger workflow (A) involves amplification of 1 or several regions of the genome with primers (blue arrows), analysis via capillary electrophoresis (multicolor peaks), and comparison of generally partial genome sequence between samples to infer relatedness. mNGS workflow (B) involves preparation of libraries (either RNA- or DNA-dependent on virus of interest) that include all RNA or DNA in the clinical sample (including mostly human and commensal flora). Reads can be aligned to a reference viral genome as pictured on the left. Alternatively, as pictured on the right, nonviral reads can be removed or viral reads can be binned via bioinformatic pipelines, and the reads of interest can be strung together via overlapping regions to create a de novo assembly. Either way, the end goal is to generate a complete viral genome with sufficient coverage of reads across the entire sequence length for comparison between samples.
- Figure 2.
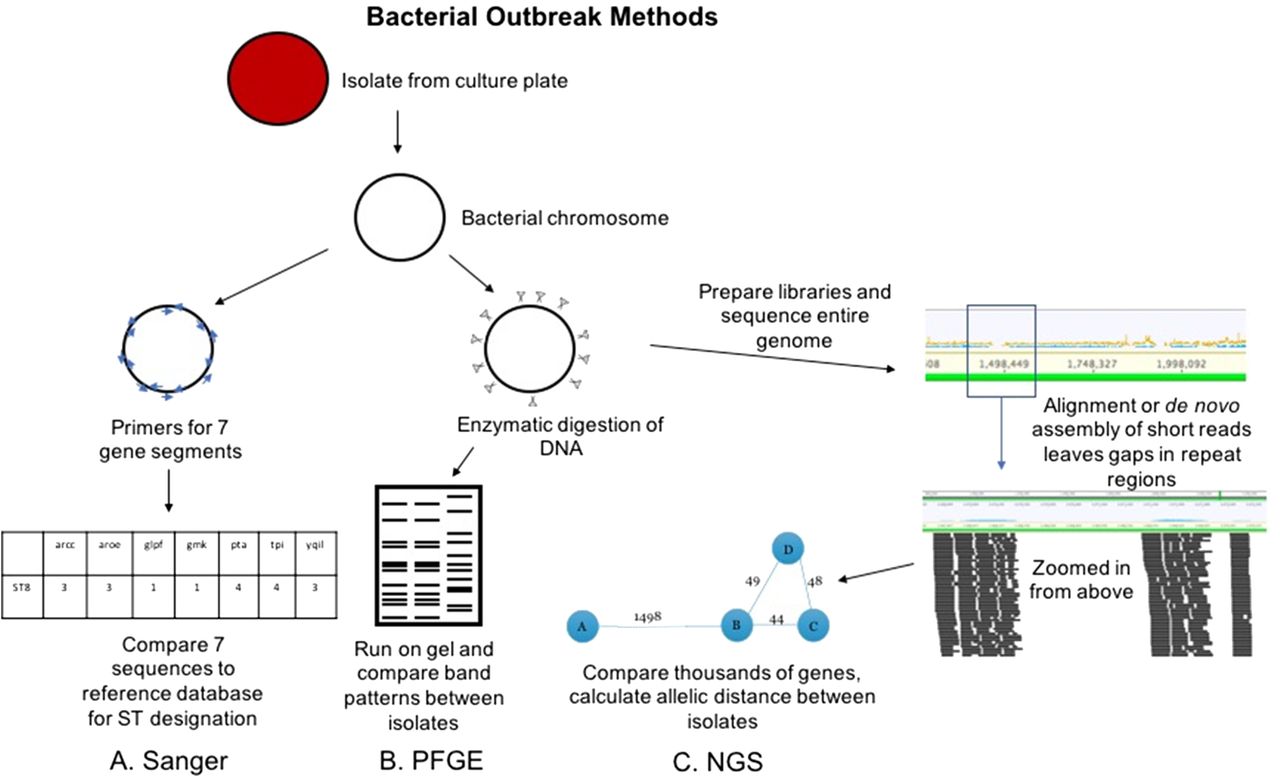
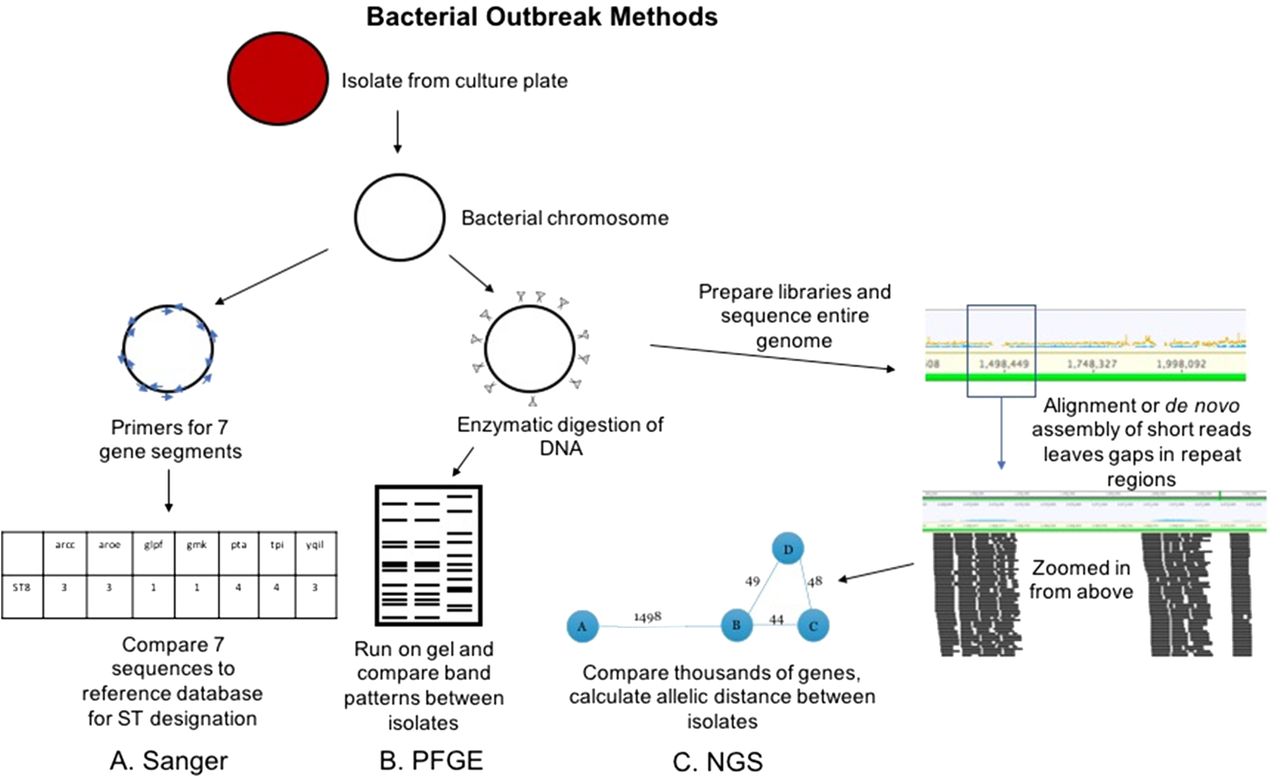
Conventional and NGS methods for bacterial outbreak investigation. The Sanger workflow (A) involves amplification of 7 gene regions with primers (blue arrows), analysis via capillary electrophoresis (like Figure 1), and comparison of the sequences to known sequence types (STs). PFGE workflow (B) involves enzymatic cutting of the bacterial chromosome followed by gel electrophoresis that alternates pulses of current in various directions to separate DNA fragments at higher resolution. The band patterns are compared between isolates to determine relatedness. NGS workflow (C) involves preparation of libraries from fragmented chromosomal DNA. Reads are assembled (either aligned or de novo as in Figure 1) to form contiguous sequences (contigs). From these contigs, thousands of genes or alleles are compared for similarity between isolates. An allelic distance, or number of alleles that are different between isolates, is calculated, and this can be visualized as the bubble and line plot shown above.
- Figure 3.
Normalized adenoviral reads are higher for samples with less background nucleic acid when adjusted for viral titer. The percent of adenoviral reads normalized to the total number of NGS reads is displayed on the x-axis. The adenovirus crossing cycle threshold (surrogate for viral titer) by real-time PCR is displayed on the y-axis. Samples with lower background human nucleic acid are in red and blue (eye and environmental samples, respectively), whereas samples with high human background are in green (respiratory samples). Mean CT -adjusted percent AdV per total reads was 5.87 times higher for eye specimens compared with respiratory specimens and was significantly different by ANOCOVA analysis (p = 0.017). CT, real-time PCR crossing cycle threshold.






{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- LEARNING OBJECTIVES
- ABSTRACT
- INTRODUCTION
- HOW DOES NGS WORK FOR WHOLE VIRAL GENOME SEQUENCING?
- CLINICAL APPLICATIONS OF NGS FOR VIRAL OUTBREAK INVESTIGATIONS
- HOW DOES NGS WORK FOR WHOLE BACTERIAL GENOME SEQUENCING?
- CLINICAL APPLICATIONS OF NGS FOR BACTERIAL OUTBREAK INVESTIGATIONS
- OTHER CONSIDERATIONS FOR BACTERIAL WGS
- WORKFORCE NEEDS FOR NGS IN OUTBREAK INVESTIGATION
- SUMMARY
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- cgMLST - core genome MLST
- GUI - graphical user interface
- MLST - multilocus sequence typing
- mNGS - metagenomic NGS
- MRSA - methicillin-resistant Staphylococcus aureus
- NGS - next-generation sequencing
- PCR - polymerase chain reaction
- PFGE - pulsed field gel electrophoresis
- SNV - single-nucleotide variation
- ST - sequence type
- WGS - whole-genome sequencing
- whole-genome sequencing
- outbreak investigation
- next-generation sequencing
- metagenomics