


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Michael A. Nardi
, New York University School of Medicine, michael.nardi{at}nyumc.org
LEARNING OBJECTIVES
1. To discuss emicizumab, including its mechanism of action and its importance in the treatment of hemophilia A.
2. To discuss how and to what extent emicizumab effects the accuracy of conventional coagulation tests.
3. To discuss the use of unconventional global coagulation tests as alternatives to assess the quantitation and effectiveness of emicizumab.
ABSTRACT
Emicizumab, a novel therapeutic agent for patients with hemophilia A (HA), is a humanized, asymmetric, bispecific, IgG4 monoclonal antibody designed to mimic the function of activated factor VIII (FVIII). Clinical studies have demonstrated its utility in the successful treatment of HA patients with and without anti-FVIII alloantibodies. The conventional methods currently used for monitoring management of HA will be discussed along with why they may be inappropriate for patients receiving emicizumab. Alternative methods (ie, global testing) will be presented and discussed in the context of emicizumab. With the increasing availability of treatment with emicizumab, it is important the clinical laboratory understands the mechanism, interaction with conventional coagulation assays, and alternative methods for its monitoring. This review describes and discusses the different assays used to measure the effectiveness of emicizumab as well as monitor FVIII activity in patients with HA.
- APCC - activated prothrombin complex concentrate
- APTT - activated partial thromboplastin time
- BPA - bypassing agent
- Ca2+ - calcium
- CSA - chromogenic substrate assay
- EGF - epidermal growth factor
- ELISA - enzyme-linked immunosorbent assay
- Fab - antigen-binding fragment
- FDA - Food and Drug Administration
- FIX - factor IX
- FIXa - activated factor IX
- FVIII - factor VIII
- FVIIIa - activated factor VIII
- FX - factor X
- FXa - activated factor X
- HA - hemophilia A
- OSA - one-stage clotting assay
- PL - phospholipid
- rhFVIIa - recombinant human activated factor VII
- ROTEM - thromboelastometry
- TEG - thromboelastography
- TF - tissue factor
- TGA - thrombin generation assay
INTRODUCTION
Hemophilia A (HA) is an X-linked hereditary bleeding disorder associated with bleeding into joints and muscle tissues and is characterized by absent or reduced quantity or dysfunction of coagulation factor VIII (FVIII). Administration of replacement factor concentrate with either plasma-derived or recombinant FVIII concentrates forms the standard treatment in patients with severe HA (<1% factor activity), with nearly all HA boys in the United States, Canada, Australia, and Northern Europe receiving prophylactic treatment, resulting in reduced morbidity and mortality compared with on-demand therapy. Such replacement therapy has successfully changed the natural history of severe HA. Previously, such patients experienced frequent bleeding events throughout life that significantly reduced quality of life and shortened lifespan. Now, patients adherent to a regimen of regular replacement with concentrates of FVIII can essentially avert all but a small number of bleeding events and experience a near-normal lifespan and quality of life.1⇓-3
Despite these benefits, approximately 50% of severe HA patients have an annual bleed rate of more than 2 despite regular replacement of the missing clotting factor administered as prophylaxis.3⇓⇓-6 Achieving optimal outcomes is hindered by lack of adherence to the FVIII dosing regimen, which requires frequent, intravenous injections of concentrates of FVIII7,8 and achieving circulating FVIII levels sufficient to prevent bleeding, including with physical activity.9 Even if adherence to a prophylaxis regimen is optimal, bleeding still occurs, resulting in long-term morbidity primarily from chronic synovitis and the development of debilitating joint disease.6
Significant barriers still remain for the treatment of patients with HA. Extended half-life FVIII products (half-life ~1.5-fold of FVIII) have been developed to reduce the frequency of intravenous infusions; however, treatment is still required twice per week.10 Regardless of the product being administered, drawbacks, such as high cost and complications associated with securing venous access, particularly in infants, continue. The most feared and clinically significant complication of factor replacement is the development of alloantibodies directed against the exogenous FVIII molecules.11,12 Inhibitors to FVIII occur in approximately one-third of those with severe HA, typically within the first 20 exposures to exogenous FVIII early in the first 2–3 years of life.13 Most importantly, they render factor replacement therapy ineffective, exposing patients to an unacceptably high risk of morbidity and mortality due to uncontrollable bleeding.14
These important considerations have led the way to developing alternative approaches for long-term prophylaxis of HA patients with or without inhibitors. Among these approaches is the development of a new class of therapeutic agents not based on coagulation factors that act by enhancing coagulation (emicizumab) or inhibiting anticoagulant pathways (fitusiran and concizumab—under development and being tested in clinical studies).15 These drugs require methods other than the conventional coagulation tests for monitoring. Laboratory evaluation of hemophilia will, therefore, undergo dramatic changes in the near future.16,17
This review will focus on the novel drug emicizumab, which is recently Food and Drug Administration (FDA) approved for the treatment of HA patients without or with inhibitors.
EMICIZUMAB
Development of Emicizumab
Emicizumab (Hemlibra, ACE910, RG6013, RO5534262, created by Chugai Pharmaceutical Co, Ltd and codeveloped by Chugai, Hoffmann-LaRoche and Genetech) is a humanized, asymmetric, bispecific, IgG4 monoclonal antibody designed to mimic the function of activated
FVIII (FVIIIa), which may be missing or defective due to a gene mutation in the F8 gene resulting in HA. Its development is a technological feat that is briefly described here. Kitazawa et al published in 201218 the compilation of nearly a decade of development of a bispecific antibody with one antigen-binding fragment (Fab) unit binding the active enzyme factor IX (activated factor IX [FIXa]) and the other binding the zymogen factor X (FX), uniting the coagulation factors together in the proper conformation for activation of FX. This was accomplished by developing 200 anti–factor IX (FIX) and 200 anti-FX Fab units and combining them to create a library of 40 000 combinations. The antibody with the combination of an ideal low binding affinity and superior activity in activated factor X (FXa) production in the setting of clot formation was selected. This candidate antibody was tolerated at high doses in animal models and corrected the hemostatic defect in an HA cynomolgus monkey model.19
Mechanism of Action
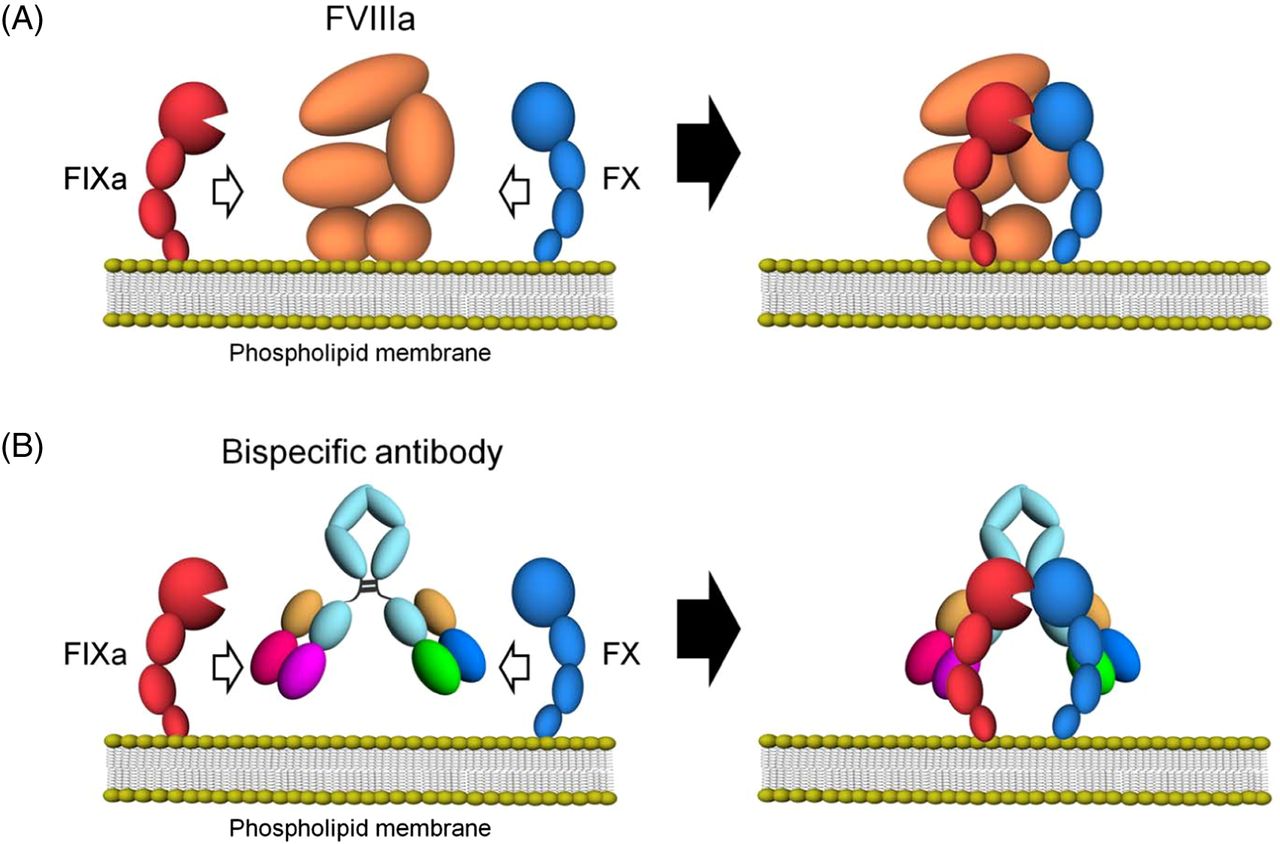
Emicizumab, through its bispecific monoclonal antibody structure, binds via one arm to the epidermal growth factor (EGF)-like domain 1 of FIX/FIXa and the other arm to the EGF-like domain of FX/FXa, mimicking the cofactor activity of FVIIIa. This bridging leads to a significant acceleration of FIXa-mediated FX activation and ultimately to increased thrombin generation20 (Figure 1). Despite the similarity in function between FVIII and emicizumab, recent studies have demonstrated differences in binding affinity and disruption of normal hemostatic regulation,21 which have implications for pharmacokinetics and dosing consideration for emicizumab prophylactic usage. The functional mechanisms of emicizumab have recently been reviewed.22,23

Schematic illustrations of cofactor actions of FVIIIa and a bispecific antibody promoting the interaction between FIXa and FX. (A) FVIIIa forms a complex with FIXa and supports the interaction between FIXa and FX through its binding ability to both factors on the phospholipid membrane. (B) A bispecific antibody binding to FIXa and FX would promote the interaction between FIXa and FX and exert FVIII mimetic activity on the phospholipid membrane. Reproduced under the terms of the Creative Commons Attribution license. Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, et al.20
Clinical Studies of Emicizumab
Emicizumab has undergone extensive clinical evaluation in multiple global, multicenter clinical trials that evaluated the safety, efficacy, and pharmacokinetics of emicizumab in HA patient populations, including pediatric, adolescent, and adult HA patients with and without anti-FVIII inhibitors. These studies were limited to small numbers of patients, especially those with inhibitors, and none were done in head-to-head comparisons between emicizumab and current HA therapies. They have been recently reviewed.3,24,25
Briefly, these studies have shown substantial reductions in bleeding rates as well as improvement of health-related quality of life.26⇓-28 Advantages of emicizumab are that it is administered subcutaneously rather than intravenously, weekly to monthly rather than 2–3 times per week, and can be used in patients with or without inhibitors, offering the former patients for the first time a convenient and prophylactic option. Following a loading dose of 3 mg/kg weekly for 4 weeks and a maintenance dose of 1.5 mg/kg weekly, 3.0 mg/kg every 2 weeks, or 6 mg/kg every 4 weeks, steady-state levels can be achieved without the constant peak and troughs associated with conventional FVIII replacement therapies.29 It has been speculated that any of these maintenance doses prevents spontaneous bleeding and minor trauma-induced bleeds, that is, changing the HA phenotype from severe (<1% FVIII) to mild (10%–15% FVIII).27
No anti-drug antibody was initially reported in the clinical studies; however, use of an optimized enzyme-linked immunosorbent assay (ELISA) drug immunoassay indicates antibodies were present. Of 398 patients, 14 (3.5%) tested positive and anti-drug antibodies. For 7 patients, the antibodies were classified as transient, whereas 3 patients were classified as having neutralizing potential (0.75% of the overall population).30
Early in the clinical studies, several cases of thrombotic microangiopathy, thrombosis, and death were noted in patients with anti-FVIII inhibitors or breakthrough bleeding events receiving repeated, high doses (>100 U/kg/d for >1 day) of activated prothrombin complex concentrates (APCCs). It was postulated that this treatment regimen may lead to accumulation of large doses of the substrates FIXa and FX, leading to a massive synergistic effect on increasing thrombin generation. In vitro experiments show that an analogue of emicizumab, combined with APCC, led to a 17-fold increase in thrombin generation, which is well above the normal physiologic range. It is now recommended not to administer APCC to persons treated with emicizumab unless absolutely indicated and only in an in-patient setting.31,32
Emicizumab was approved by the FDA in November 2017 for use as routine prophylaxis administered once a week to prevent or reduce the frequency of bleeding episodes in adults and children ≥12 years of age with HA with FVIII inhibitors. It was later approved by the FDA in October 2018 for use in patients with HA without FVIII inhibitors.29 The major achievement of emicizumab over other HA therapeutics is in its subcutaneous route for administration, long half-life of 28 days, and increased efficacy in bleed prevention regardless of the absence or presence of anti-FVIII inhibitors.26,33
LABORATORY TESTING
Measurement of FVIII and FVIII Inhibitors
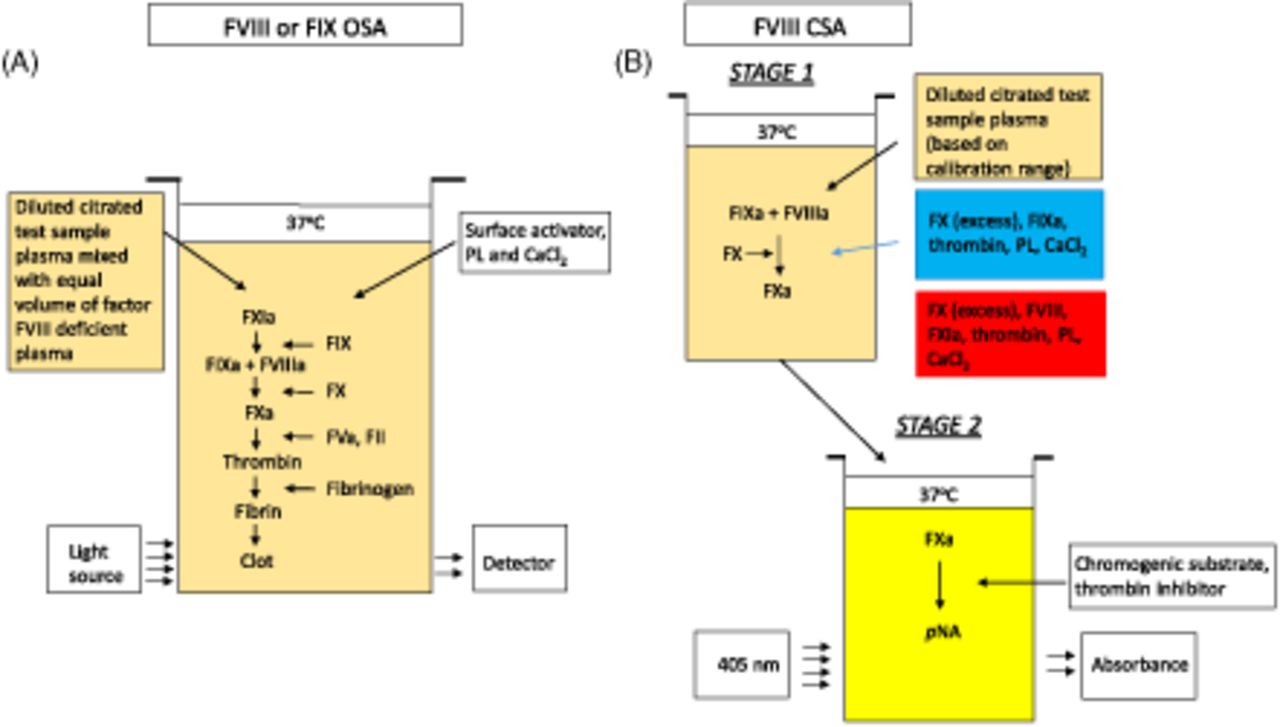
Two functional assays most commonly used for quantification of FVIII are the one-stage clotting assay (OSA) and the chromogenic substrate assay (CSA) (Figure 2). The OSA measures the ability of patient plasma to reduce the activated partial thromboplastin time (APTT) clotting time of FVIII- or FIX-deficient plasma (obtained from congenitally deficient patients or immunodepleted normal pooled human plasma). Different dilutions of patient plasma are added to deficient plasma and preincubated with an APTT reagent containing phospholipid (PL) and a contact activator (e.g., silica, ellagic acid, kaolin, polyphenols). After a set incubation time, calcium (Ca2+) chloride is added, and the time to fibrin clot formation is measured according to the analyzer’s methodology and algorithm. The result is interpolated from a calibration curve generated with a reference plasma of known FVIII and FIX concentrations.34 The large number of APTT reagents, deficient plasmas, reference plasmas, instruments, and combinations thereof results in a high level of variability.35⇓-37

Schematics of FVIII activity assays. (A) One-stage clotting assay. (B) Chromogenic substrate assay. CaCl2, calcium chloride; CSA, chromogenic substrate assay; FII, factor II (prothrombin); FIX, factor IX; FIXa, activated FIX; FVIII, factor VIII; factor VIIIa, activated FVIII; FX, factor X; FXa, activated FX; PL, phospholipid; pNA, para-nitroanaline.
The CSA for FVIII is a 2-stage assay. In the first stage, a reagent containing purified FIXa, FX, and thrombin as well as PL and Ca2+ are added in optimal concentrations to the patient’s plasma, resulting in the generation of FXa. This step does not require the extrinsic or intrinsic pathways due to the direct activation of FVIII by thrombin. In the second stage, the amount of generated FXa is measured by cleavage of an FXa-specific chromogenic substrate releasing para-nitroanaline, which causes a color change detected spectrophotometrically at 405 nm.34,38,39
Anti-FVIII inhibitors can be identified in the laboratory most commonly using the OSA but also using the CSA or an ELISA.40 The classic Bethesda assay involves normal pooled plasma as the source of FVIII being incubated in undiluted patient plasma for 2 hours at 37oC and then assayed for residual FVIII via OSA or CSA. One inhibitor unit (Bethesda Unit) is defined as the amount of patient plasma that destroys 50% of the FVIII in the mixture, corrected for the deterioration of FVIII in a control consisting of normal plasma incubated with buffer. Positive results may require further dilution of the patient plasma to be assayed to obtain an accurate result.40⇓-42 The Nijmegen modification of this assay has been shown to increase the specificity of low-titer FVIII inhibitors. The modification involves buffering the normal pooled plasma used in patient and control mixtures to pH 7.4 with imidazole buffer and using FVIII-deficient plasma in the control mixtures for preparing patient dilutions. Both adjustments maintain the pH of the reaction mixture for the 2-hour incubation and thereby stabilize FVIII in the pooled normal plasma.43 In comparison to the OSA, the CSA procedure is more standardized and offers higher interlaboratory precision in measurement of FVIII plasma activity.39
Effect of Emicizumab in Current Coagulation Laboratory Assays
Although emicizumab mimics the activity of FVIIIa, the structure as well as biochemical and functional properties of the 2 molecules are known to be different. For example, emicizumab binds to both human FIXa and FX, resulting in interference with all intrinsic pathway clotting-based assays. Also, emicizumab does not require activation by thrombin to facilitate propagation of the clotting cascade, eliminating the major rate-limiting step to fibrin clot formation, resulting in the APTT appearing to normalize at subtherapeutic emicizumab concentrations.20,21,26,46,47 Given the long half-life of emicizumab, residual interference on APTT and APTT-based assays can continue for up to 6 months after discontinuation of treatment.20
The interference analyses have shown emicizumab renders all APTT-based assays inaccurate, regardless of the reagents used. Emicizumab does not exert a similar effect on non–APTT-based clotting assays, such as thrombin time and fibrinogen assays. Chromogenic assays (e.g., protein C, antithrombin), ELISA (e.g., plasminogen), and latex immunoagglutination tests (e.g., D-dimer, free protein S, von Willebrand antigen and activity) are not influenced by the presence of emicizumab, continuing to give accurate results. There is a weak, reagent-dependent effect by emicizumab on PT and PT-based factor assays but is considered unlikely to be of clinical relevance48 (Table 1). Thus, selected conventional coagulation methods may be inappropriate for monitoring of HA patients receiving emicizumab. Alternative methods may be necessary to monitor such patients.
Effect of emicizumab on clinical coagulation assays
Measurement of FVIII and FVIII Inhibitors in the Presence of Emicizumab
Although emicizumab is effective in preventing bleeding events in HA patients, patients may require FVIII concentrates in certain situations during which it may be necessary to measure the FVIII activity level attained with the infused FVII concentrate.
As mentioned previously, emicizumab selectively binds human FIXa and FX. This can lead to falsely elevated OSA measurements of FVIII, with activities exceeding 150%. Consequently, all variations of OSAs and Bethesda assays using OSA should be avoided in the management of patients receiving emicizumab.48
An FVIII CSA utilizing bovine factors, on the other hand, would not be influenced by the presence of emicizumab, as the FIXa and FX in the reaction mixture only bind to the endogenous FVIII in the human plasma sample, thus reflecting the FVIII activity in the sample and not the coagulation potential of emicizumab. This same kit can be used to accurately measure FVIII inhibitor titers.49
In Japan, the addition of anti-idiotype monoclonal antibodies to render emicizumab inactive, eliminating its interference with APTT-based clotting assays, has been reported to accurately measure FVIII activity.50 However, the assay is presently not available in the United States.
Finally, immunoassays are not affected by the presence of emicizumab, and samples obtained during emicizumab treatment can be used in the solid-phase indirect anti-FVIII ELISA assay to detect and monitor FVIII alloantibodies.51
Measurement of Emicizumab
The current use of emicizumab does not require laboratory monitoring in the clinical setting, as demonstrated by the efficacy of emicizumab in the recent clinical studies. However, there are scenarios in which determination of the emicizumab concentration may be needed, such as breakthrough bleeding and subsequent treatment with on-demand coagulation factors, surgical procedures, or inhibitor monitoring.
In the clinical studies, emicizumab concentrations have been determined by a validated sandwich ELISA. However, this assay is prepared in-house and is not commercially available.52 A modified version of the OSA FVIII activity assay using an emicizumab-specific calibrator and plasma-based controls has been developed and commercialized in Europe (distributed by Haemochrom Diagnostica GmbH, Essen, Germany). The reagents are available in the United States but are not FDA approved. This assay follows that of the traditional FVIII OSA but is calibrated with an emicuzumab calibrator and a predilution of the patient sample (1:8) to enable a dynamic range between 10 and 100 µg/ml of emicizumab in the original plasma sample. Abstracts reporting excellent precision and reproducibility,53 significant correlation with the noncommercial ELISA method,54 and reliability in establishing whether emicizumab concentrations are within the previously determined efficacious range (30–70 µg/ml)55 have been published for this assay. This assay is recognized as the easiest and most accurate method for monitoring emicizumab concentration in plasma.
Global Testing
The ongoing clinical studies of emicizumab in severe HA patients with and without inhibitors have shown impressive reductions in annual bleeding rate compared with patients receiving standard FVIII therapy.24 Despite these impressive reductions in annual bleeding rates and the 4-week half-life of emicizumab, the best way to monitor emicizumab remains unanswered. Significant interpatient variance in emicizumab metabolism and the potential for antibody development emphasize the need for accurate laboratory monitoring.24 Since emicizumab interferes with coagulation tests that measure the intrinsic pathway, monitoring response to emicizumab therapy by the traditional coagulation tests, including the global APTT test and FVIII, therefore, is unreliable.48 There is growing interest in the use of other global assays, specifically whole-blood thromboelastography (TEG) and thrombin generation assay (TGA), to monitor the patient’s coagulation status during hemophilia therapy. Other assays have been reported, each in a single publication in the literature50,56⇓⇓-59 that will not be discussed in this review.
General Principle of TEG
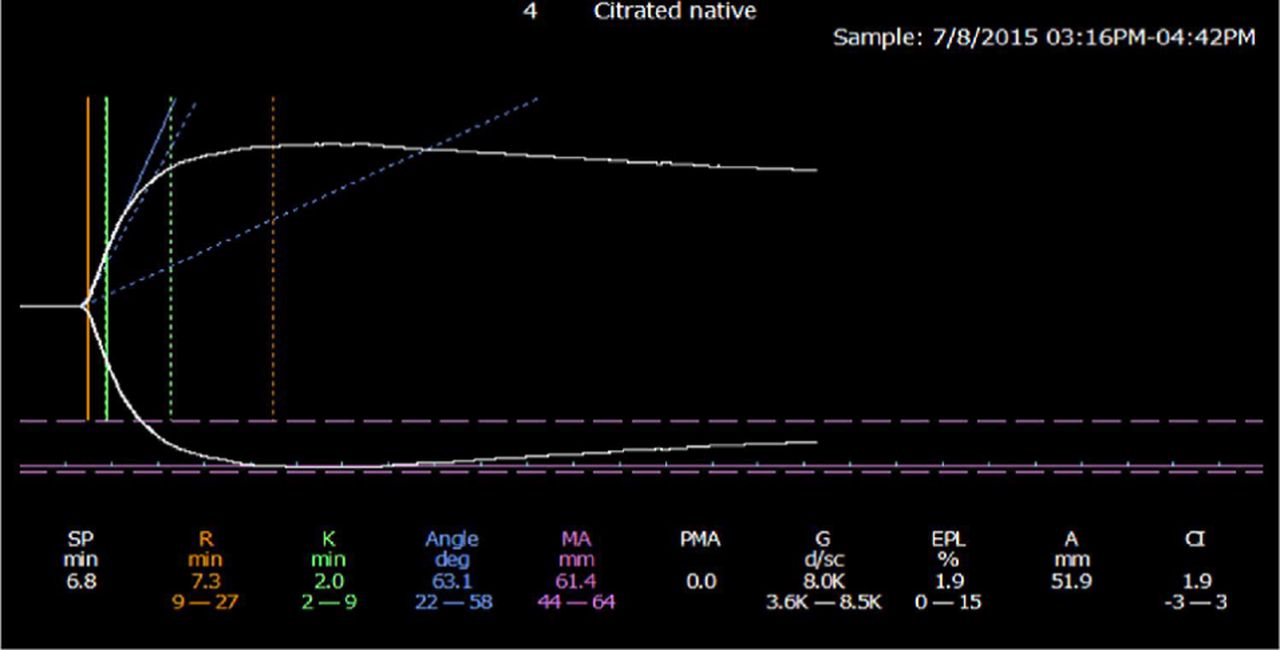
The thrombeleastographic principle, first introduced by Hellmut Hartert in 1948,60 has been modernized in the computerized version of the TEG 5000 device manufactured by Haemonetics (Braintree, MA) and the thromboelastometry (ROTEM) manufactured by Tem International (Munich, Germany). These methods measure the dynamic viscoelastic properties of whole blood during clot formation and fibrinolysis and are believed to better represent the true physiologic mechanisms of hemostasis compared with traditional coagulation assays. The devices consist of a heated cylindrical sample cup into which a pin is suspended. In the TEG 5000, the cup oscillates at ±4o45’ every 5 seconds with the pin suspended freely into the cup by a torsion wire. In the ROTEM, the cup is stationary while the pin transduction system oscillates at ±4o45’ every 6 seconds. Once the oscillation begins, the forming clot results in a physical connection by strands of fibrin between the cup and the pin, transferring the torque of the cup to the pin. The elastic strength of this connection increases gradually as the properties of the forming clot change and a mechanical electrical transducer is used to convert the rotation of the pin to an electrical signal, which is then recorded by the computer. The computer software produces both a qualitative, graphical representation (Figure 3) and quantitative parameters (Table 2), which allow one to evaluate the phases of thrombin generation, clot formation and firmness, and fibrinolysis and judge the adequacy of a patient’s coagulation system.61,62

Normal thromboelastogram (TEG). Reproduced under the terms of STM permission guidelines (2014). Chitlur M, Young G.61
Comparison of terms for TEG 5000 and ROTEM
Although thromboelastography is a general term, it mostly refers to the TEG 5000 device, whereas thromboelastometry refers to the ROTEM. For the purpose of this review, the term TEG will be used to refer to both devices.
General Principle of TGA
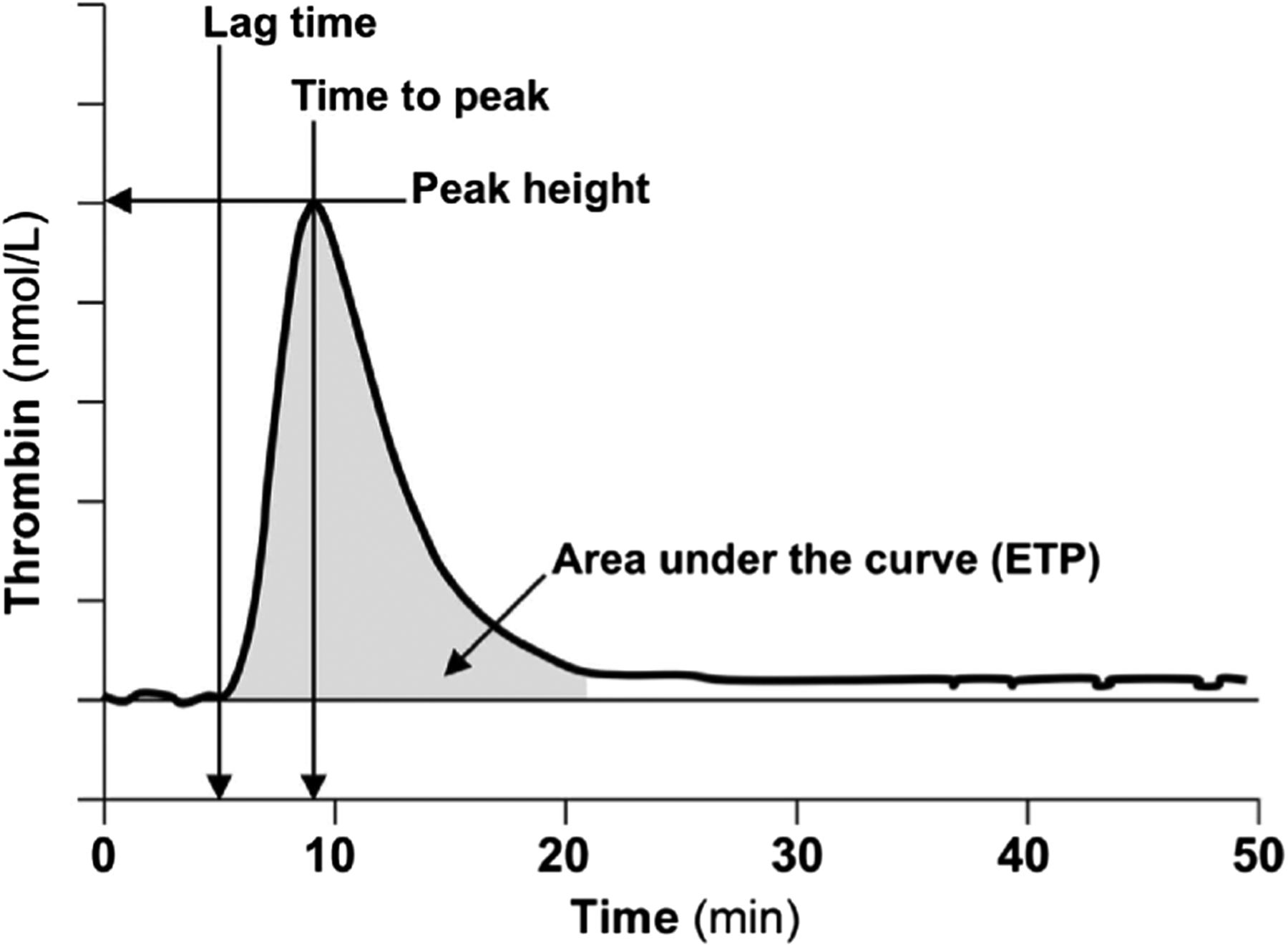
Genetic or acquired deficiency of thrombin, the key enzyme involved in maintaining hemostasis, results in a bleeding diathesis, and excessive production of thrombin is associated with a thrombotic risk. TGAs allow for measurement of the amount and rate of thrombin generated and, therefore, can be used to evaluate the patient’s overall hemostatic capacity. First described in 1953 by Macfarlane and Biggs,63 the TGA assay has been modified significantly over the years to make the test more practical using chromogenic or fluorescent thrombin substrates, allowing continuous measurement of thrombin activity in plasma. Tissue factor (TF), PLs, and Ca2+ are added to the sample plasma to induce thrombin formation. The final thrombin generation curve (thrombogram) is characterized by the lag phase (coagulation initiation), thrombin burst leading to peak thrombin concentration (acceleration and propogation), and reduction of thrombin generation due to the presence of natural anticoagulants (termination). Parameters that are reported include the lag time, peak time, peak height, and area under the curve (or endogenous thrombin potential) (Figure 4).62,64 Since TGA measures thrombin generation taking into account the function of other procoagulant and anticoagulant factors in the sample, it is considered to provide a more physiologic evaluation of the clotting system.

Typical thrombin generation assay (TGA) with relevant parameters. Reproduced with permission from the American Association for Clinical Chemistry. Tripodi A.63
Performance issues and Limitations of TEG and TGA
The 2 assays differ in a number of important ways. First, although both assays can accommodate whole-blood, platelet-rich, and platelet-poor plasma, the vast majority of studies done thus far with each device have used whole blood for TEG and platelet-poor plasma for TGA. Second, the TEG demonstrates the dynamics of clot formation overtime, whereas the TGA demonstrates thrombin generation over time. The advantages of TEG are that it uses whole blood and measures clot formation, which comes closer to the idealized coagulation assay, whereas TGA measures a surrogate marker for clot formation. The main advantage of the TGA is that by using platelet-poor plasma, samples can be stored, allowing for testing to be done at any time and in an onsite or reference laboratory, whereas TEG assays must be done within 2 hours of sampling, requiring it to be done locally.61,62,64
Although the number of devices and reagents available for use with respect to global hemostasis is somewhat limited, the use of different activators and different amounts of activators, addition or not of contact factor inhibitors, and addition or not of fibrinolytic reagents has led to considerable confusion in the literature and challenges when comparing different results from different studies. The Scientific and Standardisation Subcommittee on FVIII, FIX, and Rare Coagulation Disorders of the International Society on Thrombosis and Haemostasis recently published recommendations regarding standardization of the approach for TEG and TGA in hemophilia.65,66
TEG and TGA Monitoring of HA Patients on Emicizumab Therapy
Both in vitro and in vivo studies have been reported demonstrating the ability of TEG to predict clinical response to the bypassing agents (BPAs), APCC and recombinant human activated factor VII (rhFVIIa), in HA patients with inhibitors and to improve clinical performance of rhFVIIa during surgery.67⇓-69 Similarly, differences in TGA results to in vitro addition of FVIII or normal plasma to plasma samples from patients with HA- or FVIII-deficient plasma have been shown, which may provide relevant clinical information to guide prophylaxis as well as surgical management of patients with HA.70,71 TGA has also been used to safely guide therapy with multiple BPAs for the management of bleeding in HA patients with inhibitors.72,73
With the information obtained in these early studies, the use of TEG and TGA to monitor hemostasis in HA patients was done in the emicizumab clinical trials. Nonactivated (Ca2+-triggered) TEG has shown samples spiked with emicizumab ex vivo have improved hemostatic function in a dose-dependent manner, irrespective of the presence of FVIII inhibitors, and has been shown to be more informative in terms of global coagulation response to emicizumab than other activators (such as kaolin, TF, etc) with TEG. However, there is some evidence to suggest analyzing samples spiked with emicizumab ex vivo overestimates hemostatic function, and further research into the accuracy of TEG in emicizumab-treated patients is needed.74 TGA shows a linear, dose-dependent increase in thrombin generation in the presence of emicizumab and can be used to monitor global hemostatic response in patients treated with emicizumab.75 TGA has also been shown to monitor the cumulative effect of APCC or rhFVIIa with FVIII inhibitors and can be used as a tool to individually tailor BPA therapy. This is of particular interest for patients treated with BPAs for the treatment of breakthrough bleeding events, as in the various clinical trials.26,76
The results of an in vitro evaluation of clot formation and thrombin generation using TEG and TGA, respectively, for combinations of a sequence-identical analog of emicizumab with APCC or rhFVIIa have been reported. The analog in combination with APCC resulted in a 17-fold increase in thrombin generation over the analog alone, producing a probable hypercoagulable state that was confirmed by TEG. The analog in combination with rhFVIIa also produced an increase in thrombin generation, but it did not reach the normal range. These data suggest that considerable additional research is still warranted.31
Three case reports describing the use of global assays to help guide BPA treatment in 4 HA patients with anti-FVIII inhibitors have recently appeared in the literature. One report performed TEG and TGA for 2 HA patients after 1 and 2 months of treatment, reporting TEG showed complete correction, whereas TGA showed marked improvement, but only one-third that of a healthy volunteer, consistent with a mild hemophilia phenotype.75 The other two reports used in vitro TEG and TGA testing of the patient’s plasma with different concentrations of APCC or rhFVIIa to determine which BPA to use as well as to inform about its ideal dose.77,78 In both reports the patients were successfully supported during surgical procedures.
CONCLUSION
Emicizumab is a novel therapeutic antibody that has been shown in clinical studies to be efficacious in the treatment of HA with and without inhibitors. The benefits of emicizumab include subcutaneous administration; a half-life of 28 days allowing for reduced dosing frequency, as infrequent as monthly; greater prophylactic efficacy; and benefits irrespective of the presence of anti-FVIII inhibitors.
Its introduction into the clinical practice of hemophilia treatment requires adjustments in clinical laboratories that monitor FVIII activity and anti-FVIII inhibitor levels. Such laboratories must understand that the structural and functional differences between emicizumab and FVIII mean that many of the conventional assays used to monitor conventional HA treatment are no longer accurate when emicizumab is present in the sample. Specifically, emicizumab, even at subtherapeutic concentration, results in a shortened APTT, rendering the most commonly used APTT-based coagulation assays inaccurate. For example, when samples from patients receiving simultaneous emicizumab and conventional FVIII replacement products are referred for FVIII assessment and/or inhibitor screening/titration, a chromogenic FVIII assay utilizing bovine reagents would be required for the accurate measurement of FVIII activity. Thus, it is imperative the laboratory personnel receive accurate information about the patient’s therapy treatment. Communication between the ordering physician and clinical laboratory is essential for delivery of accurate and appropriate results.
To measure the concentration and activity of emicizumab, a modified FVIII OSA calibrated against emicizumab can be used in the clinical setting; however, this assay is presently not available in the United States.
There is increased interest in global assays, such as TEG and TGA, for monitoring coagulation potential in HA patients receiving emicizumab. Both assays have been reported to provide clinically relevant information. The TGA has been shown to be useful in predicting an appropriate therapeutic concentration of the common BPAs, APCC and rhFVIIa. Further research, however, is necessary to confirm the accuracy of TEG and TGA in the presence of emicizumab. The ability to measure emicizumab activity and concentration in the coagulation laboratory may require a shift from the functional assays of the past to assays that are specifically tailored for use in the patients receiving emicizumab.
- Received December 17, 2019.
- Revision received March 6, 2020.
- Accepted April 30, 2020.
American Society for Clinical Laboratory Science
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.
- 45.
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- APCC - activated prothrombin complex concentrate
- aPTT - activated partial thromboplastin time
- BPA - bypassing agent
- Ca2+ - calcium
- CSA - chromogenic substrate assay
- EGF - epidermal growth factor
- ELISA - enzyme-linked immunosorbent assay
- Fab - antigen-binding fragment
- FDA - Food and Drug Administration
- FIX - factor IX
- FIXa - activated factor IX
- FVIII - factor VIII
- FVIIIa - activated factor VIII
- FX - factor X
- FXa - activated factor X
- HA - hemophilia A
- OSA - one-stage clotting assay
- PL - phospholipid
- rhFVIIa - recombinant human activated factor VII
- ROTEM - thromboelastometry
- TEG - thromboelastography
- TF - tissue factor
- TGA - thrombin generation assay
- hemophilia A
- emicizumab
- Hemlibra
- global assays
- thromboelastography
- thrombin generation assay