


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Billie Ketelsen
, Oakland University, billie.ketelsen{at}gmail.com
LEARNING OBJECTIVES
1. Explain normal functioning of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13), in the plasma.
2. Describe the abnormal functioning of ADAMTS-13 in congenital and acquired thrombotic thrombocytopenic purpura (TTP).
3. Describe the mechanism of transfusion therapy for TTP.
ABSTRACT
Thrombotic thrombocytopenic purpura (TTP) is a disease that is classified by abnormal functioning of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13), protease. ADAMTS-13 protease impairment can be caused by genetic mutations at the gene level or through autoantibodies that are formed within the circulation. Congenital mutations account for about 5%–10% of the TTP population, whereas the acquired version is more common. The acquired version of TTP is caused by inhibitory and noninhibitory autoantibodies that affect the ADAMTS-13 protease. Both congenital and acquired TTP are treated through transfusion therapy with therapeutic plasma exchange (TPE). TPE is used to remove the autoantibodies and any mutated ADAMTS-13 proteases in the circulation while providing the addition of normal functioning ADAMTS-13 to the circulation.
- ADAMTS-13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif - member 13
- CPP - cryoprecipitate-poor plasma
- CUB - complement C1r/C1s, Uegf, Bmp1
- FFP - fresh frozen plasma
- FP24 - plasma frozen within 24 hours after phlebotomy
- IgG - immunoglobulin G
- IgM - immunoglobulin M
- LDH - lactate dehydrogenase
- TP - thawed plasma
- TPE - therapeutic plasma exchange
- TSP - thrombospondin
- TTP - thrombotic thrombocytopenic purpura
- ULVWF - ultra-large von Willebrand factor
- VWF - von Willebrand factor
INTRODUCTION
Thrombotic thrombocytopenic purpura (TTP) is the common term for patients who present with thrombocytopenia and microthrombi formation in the small capillaries. These thrombotic microangiopathies may be in seen adults with or without predominant neurologic symptoms. TTP may occur at any age; however, the peak incidence is in the third decade of life.1 TTP clinically presents similarly to other thrombotic microangiopathies and often requires a diagnosis through exclusion. Classic TTP shows symptoms of hemolytic anemia, thrombocytopenia, neurologic symptoms, and fever. Early intervention is essential for patients presenting with TTP because untreated mortality rates are approximately 90%. Treated mortality rates decrease drastically to 10%–20%.2 At the vascular level, TTP is a disease with a deficiency of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13), that results in accumulation of ultra-large von Willebrand factor (ULVWF), which drives activation of intravascular platelets, resulting in microvasculature thrombi.3 ULVWF multimers are formed in large strings in the endothelium and are anchored to the cell wall. These multimers are extremely adhesive to platelet glycoproteins, causing platelet aggregation.4
ADAMTS-13
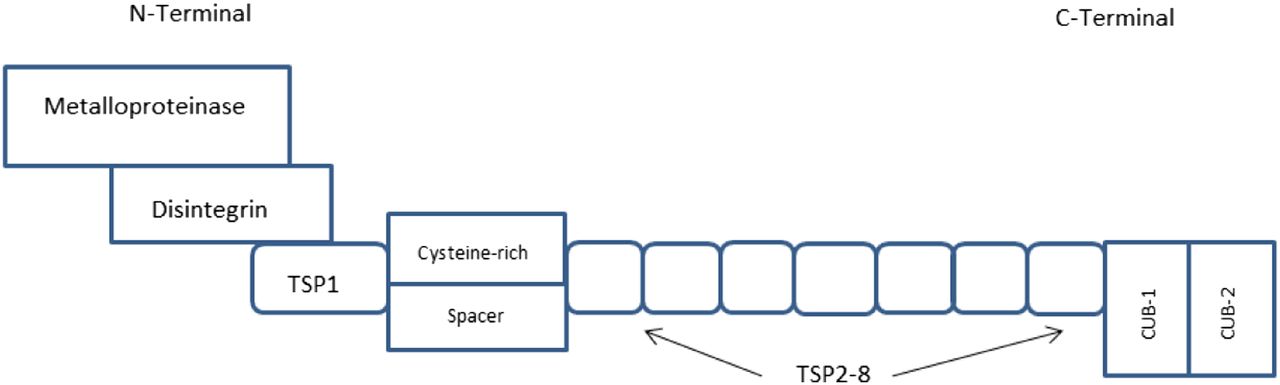
ADAMTS-13 is encoded by the ADAMTS-13 gene located on chromosome 9q34, where it is composed of 29 exons and covers approximately 37 kilobases (kb) on chromosome 9.5,6 The basic structure of ADAMTS-13 includes a metalloproteinase (proteolytic domain), thrombospondin-1 (TSP-1)-like domains for a total of 8 domains, a cysteine-rich/spacer motif, and 2 complement C1r/C1s, Uegf, Bmp1 (CUB) structures (peptide-containing domains). The N-terminal includes the metalloproteinase, disintegrin, TSP-1, and the cysteine-rich/spacer. The C-terminal includes the TSP-2–TSP-8 domains and the 2 CUB structures (Figure 1).4 ADAMTS-13 is synthesized in the hepatic stellate cells of the liver and, to a lesser extent, endothelial cells. ADAMTS-13 is secreted into circulation as an active enzyme different from other ADAMTS proteases and results in a precursor protein that is 1427 amino acids in length (approximately 145 kDa).5,6 Further processing of the ADAMTS-13 transcripts is complex and varied. In the plasma, there have been several forms of ADAMTS-13 transcripts isolated, including 170 kDa, 160 kDa, and 120 kDa proteins that contain an N-terminal amino acid sequence that is identical in all forms.5 In an active state, ADAMTS-13 is a folded protein where the CUB domains interact with the spacer region of the proteinase.7

Domain structure of ADAMTS-13. ADAMTS-13 is composed of a metalloproteinase (proteolytic domain), TSP-1–like domains for a total of 8 domains, a cysteine-rich/spacer motif, and 2 CUB structures (peptide-containing domains). The N-terminal domain of ADAMTS-13 is composed of the metalloproteinase, disintegrin, TSP-1, and the cysteine-rich/spacer domains. The C-terminal domains include TSP-2–TSP-8 and the 2 CUB structures.
When normal functioning is observed, ADAMTS-13 protease cleaves von Willebrand factor (VWF) between tyrosine 1605 and methionine 1606 in the A2 domain of the VWF protein; this results in a mature VWF protein. The main target of this cleavage is the ULVWF molecules that are the precursor of mature VWF. During high shear events, ULVWF unfolds, exposing the A2 site to the ADAMTS-13 protease for cleavage. Deficiency in ADAMTS-13 protease allows for ULVWF to remain uncleaved in the vasculature, resulting in the aggregation of microclots. ADAMTS-13 protease deficiencies can be seen in cases of a genetic mutation (homozygous or heterozygous) or autoimmune inhibitors of the circulating protease.8 Under normal conditions, ADAMTS-13 circulates at a concentration of 1 µg/mL with a plasma half-life of 2–3 days.6
GENETIC MUTATIONS AND CONGENITAL TTP
When a mutation occurs at the gene site, the result is an absence or deficiency of ADAMTS-13 protease.1 At the time of publication, there were over 70 identified mutations in the ADAMTS-13 gene, resulting from various missense, nonsense, frame shift, or splice site mutations.2 Each mutation class affects a different portion of the gene’s coding region, impairing different variations of gene synthesis, secretion, or proteolytic activity.6 The result of a genetic mutation results in congenital (familial) TTP and rarely has an autoimmune component to the disease presentation.4
Congenital TTP is a relapsing disorder in which patients have less than 5%–10% of normal plasma ADAMTS-13 levels, and it accounts for 5% of TTP cases. For these patients, their ADAMTS-13 levels remain in that range during and between thrombotic episodes. Congenital TTP is caused by an autosomal recessive inheritance of homozygous or double heterozygous mutations in the ADAMTS-13 gene. TTP episodes usually begin in infancy/childhood and continue throughout the patient’s life.4 Compound heterozygous mutations account for 65% of familial cases, with the remaining 35% caused by homozygous mutations in the gene’s coding region.
INHIBITORS AND ACQUIRED TTP
Autoimmune inhibitors of the ADAMTS-13 protease are responsible for 95% of all TTP cases, and that inhibition is known as acquired TTP.6 The antibody response includes inhibitory and noninhibitory antibodies. The inhibitory antibodies block the proteolytic activity of ADAMTS-13 toward VWF, and the noninhibitory antibodies aid in increasing ADAMTS-13 clearance from the patient’s circulation. The principal antibody class for acquired TTP is immunoglobulin G (IgG) with a minor class of immunoglobulin M (IgM) and immunoglobulin A antibodies present in cases.6,8 Of the IgG class molecules, IgG1 and IgG4, molecules that differ only in their Fc regions, are the predominant IgG antibodies present. IgG1 antibodies are found most commonly in the beginning stages of acquired TTP and with the first acute episode of the disease, whereas IgG4 antibodies can be found in cases of prolonged stimulation and are common in later stages of acquired TTP.9 Although IgM antibodies are less common, one study showed that the presence of IgM antibodies in the circulation resulted in an overall increase (35%–79%) in ADMATS-13 activity as compared with less than 10% activity in the presence of IgG antibodies.10
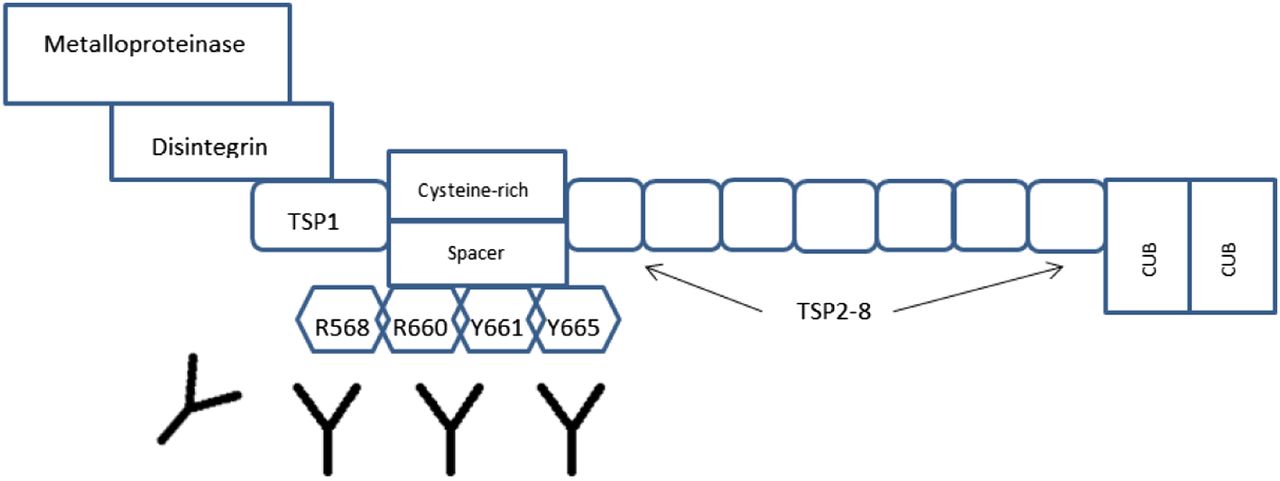
Acquired TTP patients show transient inhibition of ADAMTS-13 with levels less than 5%–10% during acute thrombotic episodes, and etiologies are mostly unknown.4,11 During nonthrombotic events, ADAMTS-13 activity may be in a normal range because of the natural decrease of inhibitors within the circulation.11 During thrombotic events, autoantibodies formed will show affinity toward the specific metalloprotease and spacer domains of the ADAMTS-13 transcript located on the N-terminal end.11 In these events, ADAMTS-13 appears in a more open configuration, exposing cryptic epitopes in the spacer region that are the site for autoantibody attachment. These epitopes have been described as R568, R660, Y661, and Y665 in multiple studies and are found in the spacer domain of the protease (Figure 2).7 This area of the gene is significant for proper protease function.11 The determination between inhibitory and noninhibitory antibodies in acquired TTP cases can only be determined through laboratory testing.

Cryptic epitopes in the spacer domain that are targeted by autoantibodies to ADAMTS-13.
TRANSFUSION THERAPY FOR ACQUIRED TTP
Treatment of both congenital and acquired TTP is the same: therapeutic plasma exchange (TPE). The purpose of TPE is to remove molecules from the plasma that are inhibitory or replace normal substances that may be missing from the circulation. The molecules that can be removed include a variety of high-molecular weight complexes, antibodies, and proteins, whereas normal substance replacements can include coagulation factors or enzymes. In TTP, the goal of TPE is to perform both indications: to remove inhibitory antibodies or nonfunctioning ADAMTS-13 molecules while replacing normal functioning ADAMTS-13.3 TPE is a lengthy procedure that includes removing defective plasma from the circulation and replacing the volume with normal plasma, and it is a concurrent exchange of a person’s total plasma volume. This process occurs by removing whole blood from the circulation, extracting the plasma, and returning the remaining blood products with the new plasma back to the circulation.
Active ADAMTS-13 can be found in many different plasma products, including fresh frozen plasma (FFP), plasma frozen within 24 hours after phlebotomy (FP24), or cryoprecipitate-poor plasma (CPP). Once the frozen plasma product has been thawed, it is considered thawed plasma (TP). Plasma products can be prepared from a whole blood donation or a plasma apheresis procedure. Once collected, there are different storage and preparation techniques that can be used. In most cases, the plasma product is frozen quickly to maintain coagulation factor activity. FFP is the name given to a plasma product that has been frozen within 8 hours of collection and processing. FP24 is a plasma product that has been frozen within 24 hours of collection and processing. CPP is the plasma byproduct of cryoprecipitate preparation. When cryoprecipitate is removed from plasma, the remaining plasma is frozen within 24 hours to create CPP. Some coagulation factors are decreased in CPP compared with the other plasma products available. All frozen plasma products have a shelf life of 12 months once frozen and cannot be used clinically until they are thawed. TP is created when frozen plasma of any form is thawed for use. TP has a shelf life of 5 days once thawed and can be used in all clinical situations. ADAMTS-13 levels are maintained at normal levels in TP for the entire 5 days.2
The half-life of infused ADAMTS-13 through plasma is 2–4 days, which is similar to the normal acting half-life of ADAMTS-13.4 TPE is used as a front-line treatment for multiple reasons: It is easily accessible, it can clear ADAMTS-13 antibodies from the circulation, and it can add normal functioning ADAMTS-13 back into the circulation. There can be adverse effects associated with TPE, as with any transfusion. Some noted adverse effects of TTP include hypocalcemia caused by the infusion of citrate with the returned blood, a burning or prickling sensation (paresthesia), and, occasionally, nausea.3 Over time, CPP may be a better option for TPE because CPP contains fewer high-molecular-weight VWF multimers than standard FFP.2
The normal course of TPE for an acquired TTP case is 5–7 days of treatment. Each day of treatment removes a portion of the autoantibodies present in the circulation, replacing the removed volume with clinically active ADAMTS-13. TPE works optimally when the antibody class is primarily IgG, found in the majority of acquired TTP cases. TPE may have to be extended if IgM antibodies are present or the platelet count does not increase above 150 000/µL.2 If IgM is the primary antibody, it will take longer to remove from the plasma because of the size of the antibody. One IgM molecule is 5 times the size of an IgG antibody.
Congenital TTP is treated on a more routine schedule if TPE allows for the removal of any ADAMTS-13 antibodies, inhibitors, or additional VWF mulitmers.3 Continual treatment of congenital TTP by TPE prevents thrombotic events. Suggested TPE protocols require 10–15 mL/kg body weight at intervals of 2–3 weeks.
The treatment of acquired TTP consists of daily TPE with a volume of 1–2 times the patient’s blood volume, approximately 40–60 mL/kg body weight. This is continued until a platelet count at or above 150 000/µL is observed for 2–3 days, the lactate dehydrogenase (LDH) normalizes, and any neurologic adverse effects subside. LDH is commonly seen elevated in thrombotic TTP episodes because of intravascular hemolysis. The red blood cells in the microcirculation release LDH in these events. LDH values will normalize as red blood cell hemolysis decreases. After stabilization in laboratory results, TPE can be administered every other day if needed.1,4 Laboratory values for a typical course of TPE show platelet counts rising and LDH values decreasing until a normalized state is observed (Table 1).
40-year-old female undergoing treatment TPE for TTP
Platelet transfusions are not a treatment option for TTP, although thrombocytopenia is present. As reviewed, in TTP cases, platelets interact with ULVWF to create microthrombi within the circulation. This can lead to a drastic decrease in platelets in circulation. The addition of new platelets through transfusion could increase the thrombotic events without removing the ADAMTS-13 inhibitor prior to transfusion.2
Presently, there are a few recombinant drug options available to aid in treatment of TTP. One new recombinant product that is being examined is caplacizumab, an anti-VWF nanobody.12
Table 1 shows the laboratory values of a 40-year-old female who presented to the emergency department with TTP symptoms. The patient is known to have acquired TTP prior to this visit. The patient underwent daily TPE for a course of 6 days at 3000 mL plasma volume the first 3 days and 3500 mL plasma exchange the next 3 days. Plasma exchange volume was appropriately dosed based on that patient’s body weight and was increased on day 4 to better her platelet response. On day 6 of treatment, the patient’s platelets had reached the appropriate threshold of above 150 billion (bil)/L, and LDH values were within normal ranges. The patient was discharged on day 7 without complication.
CONCLUSION
TTP is a disease that can be the result of a genetic mutation in or an acquired antibody to the ADAMTS-13 protease molecule. ADAMTS-13 is essential for preventing microthrombi in the circulation, and both branches of TTP change the normal functioning of ADAMTS-13. A standard treatment of TTP includes rounds of TPE to remove the antibodies and inhibitors that may be found in the circulation of patients with TTP. By removing the antibodies in circulation, ADAMTS-13 can cleave VWF as expected and prevent platelet aggregation that leads to the microthrombi.
- Received February 12, 2020.
- Revision received May 8, 2020.
- Accepted June 4, 2020.
American Society for Clinical Laboratory Science




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- ADAMTS-13 - a disintegrin and metalloproteinase with a thrombospondin type 1 motif - member 13
- CPP - cryoprecipitate-poor plasma
- CUB - complement C1r/C1s, Uegf, Bmp1
- FFP - fresh frozen plasma
- FP24 - plasma frozen within 24 hours after phlebotomy
- IgG - immunoglobulin G
- IgM - immunoglobulin M
- LDH - lactate dehydrogenase
- TP - thawed plasma
- TPE - therapeutic plasma exchange
- TSP - thrombospondin
- TTP - thrombotic thrombocytopenic purpura
- ULVWF - ultra-large von Willebrand factor
- VWF - von Willebrand factor
- ADAMTS-13
- autoantibodies
- IgG
- thrombotic thrombocytopenic purpura
- therapeutic plasma exchange