


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Erin Graf
, Children’s Hospital of Philadelphia, erinhgraf{at}gmail.com
LEARNING OBJECTIVES
1. Compare the advantages of next-generation sequencing (NGS) approaches to viral outbreak investigation over conventional Sanger sequencing.
2. Describe real-time clinical applications of NGS for viral and bacterial outbreak investigation.
3. Contrast the differences between pulsed field gel electrophoresis and Sanger sequencing multilocus sequence typing (MLST) approaches to bacterial outbreak investigation compared with NGS-based approaches, such as core genome MLST.
4. Discuss the wet bench and analytical workforce skills needed for implementation of NGS for outbreak investigation.
ABSTRACT
With the advent of next-generation sequencing technologies, genomic investigations of pathogen outbreaks are now possible at unprecedented resolution. Prior methods requiring highly pathogen-specific protocols can now be replaced with a single universal protocol for whole-genome sequencing. Clinical applications have shown improvement in our understanding of pathogen transmission dynamics from the hospital to global level. However, there are several barriers to routine implementation in clinical microbiology laboratories. These include financial support for a nonbillable test; workforce training needs, particularly for highly complex data analysis methods; and lack of Food and Drug Administration–approved methods requiring extensive laboratory validation.
- cgMLST - core genome MLST
- GUI - graphical user interface
- MLST - multilocus sequence typing
- mNGS - metagenomic NGS
- MRSA - methicillin-resistant Staphylococcus aureus
- NGS - next-generation sequencing
- PCR - polymerase chain reaction
- PFGE - pulsed field gel electrophoresis
- SNV - single-nucleotide variation
- ST - sequence type
- WGS - whole-genome sequencing
INTRODUCTION
Investigation of hospital or community-associated infectious disease outbreaks has historically required complex and pathogen-specific protocols generally limited to reference and public health laboratories. For viral outbreaks, Sanger sequencing approaches are still routinely used. These protocols include amplification of 1 or more gene regions that are unique to the specific virus being targeted followed by capillary electrophoresis (Figure 1A). Amplification of nucleic acid isolated from the supernatant of virus grown in culture is the most sensitive Sanger sequencing approach. Since viral culture is no longer routinely performed in the majority of clinical laboratories, Sanger protocols have also been successfully applied directly to clinical samples. However, the specimen matrix and the quantity of virus present both impact performance. For the former, the higher the quantity of virus, the more likely Sanger sequencing will be successful. A second round of amplification, also known as “nested PCR,” can often overcome low quantity issues but introduces the possibility of sequence bias.1 Clinical specimens can contain substances that inhibit amplification (eg, stool). In addition, the presence of human and/or bacterial flora nucleic acid can lead to cross-amplification and take away from amplification of the target viral sequence.2

Conventional and NGS methods for viral outbreak investigation. The Sanger workflow (A) involves amplification of 1 or several regions of the genome with primers (blue arrows), analysis via capillary electrophoresis (multicolor peaks), and comparison of generally partial genome sequence between samples to infer relatedness. mNGS workflow (B) involves preparation of libraries (either RNA- or DNA-dependent on virus of interest) that include all RNA or DNA in the clinical sample (including mostly human and commensal flora). Reads can be aligned to a reference viral genome as pictured on the left. Alternatively, as pictured on the right, nonviral reads can be removed or viral reads can be binned via bioinformatic pipelines, and the reads of interest can be strung together via overlapping regions to create a de novo assembly. Either way, the end goal is to generate a complete viral genome with sufficient coverage of reads across the entire sequence length for comparison between samples.
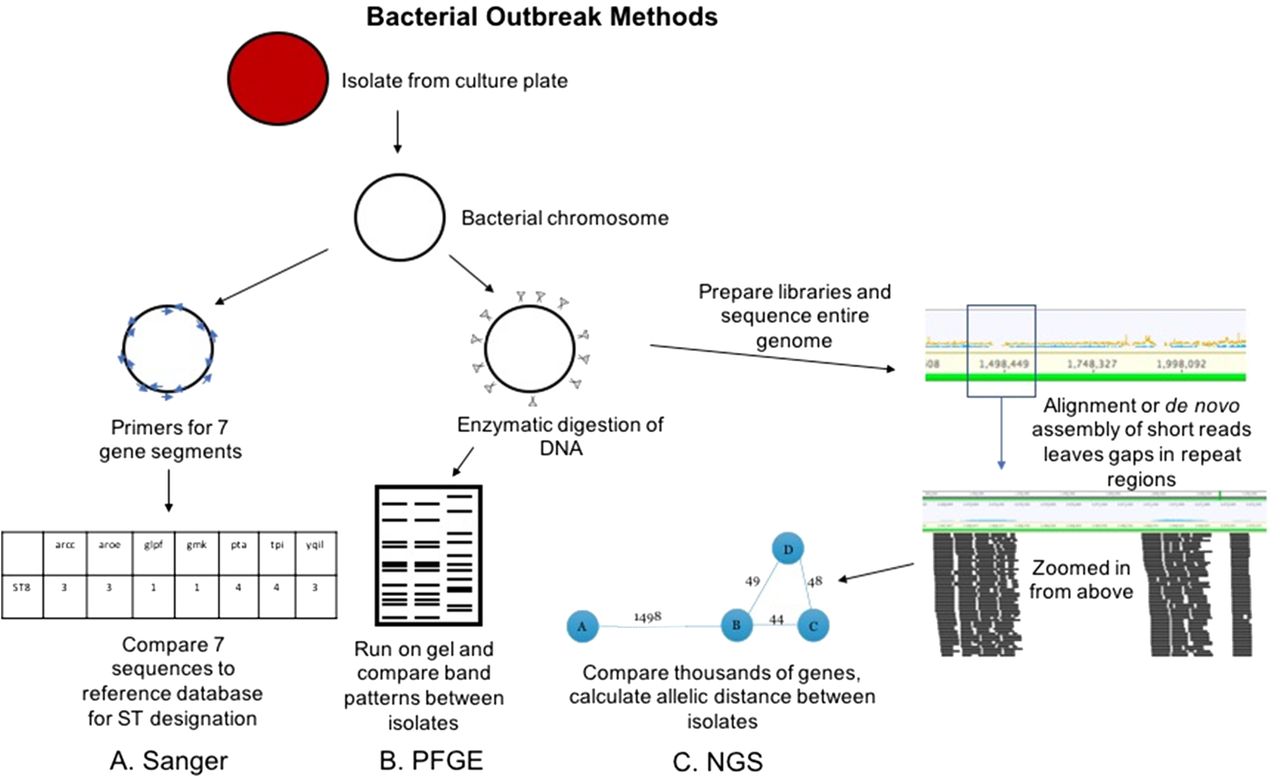
For bacterial outbreaks, there are 2 primary conventional approaches. The first is termed pulsed field gel electrophoresis (PFGE). This method uses DNA carefully extracted from isolated bacterial colonies such that large chromosomal pieces are maintained. The DNA is then sheared with specific enzymes and run on an electrophoresis gel with current applied at varying directions to allow for higher resolution of bands that may differ by small numbers of base-pairs (Figure 2B).3 Band patterns are analyzed either visually or with commercially available software and compared across isolates to look for similarities.4 The number of bands that are different between isolates is compared to published or validated criteria.5 For example, isolates that have 3 bands different would be considered closely related and possibly part of an outbreak, whereas those with 8 bands different would be considered unrelated. PFGE has been the backbone for public health investigations of food-borne outbreaks for the last 20 years.6 Sanger sequencing has also been applied to bacterial outbreak investigation (Figure 2A). Amplification and sequencing of a limited set of housekeeping genes is known as multilocus sequence typing (MLST).7 The list of genes is specific to each bacterial species and usually includes 7 genes, though there are ribosomal typing approaches that incorporate more genes. The pattern of the sequence for these genes is what determines the bacterial sequence type (ST), which can then be compared across isolates. This approach is lower resolution compared with PFGE. In other words, if isolates are different STs, one can conclude they are not related; however, if they are the same ST, they may be related or, coincidentally, the same ST without being related.
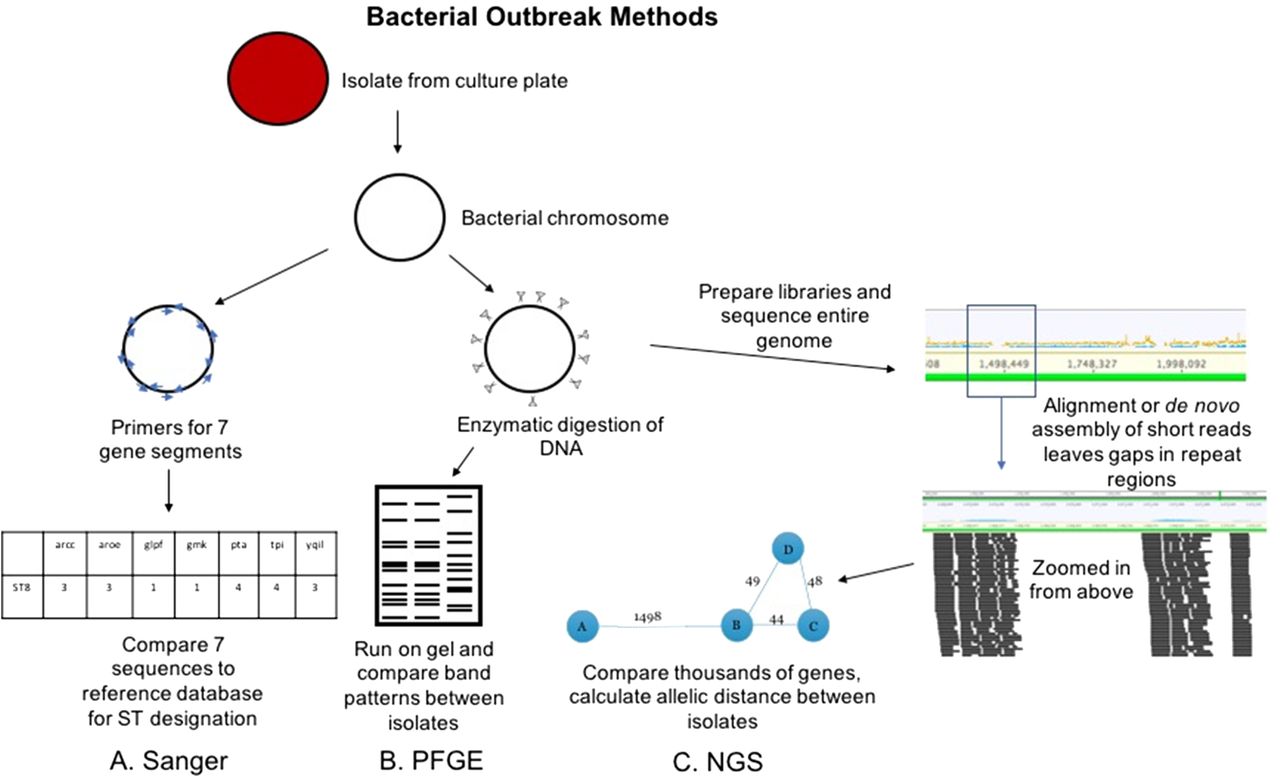
Conventional and NGS methods for bacterial outbreak investigation. The Sanger workflow (A) involves amplification of 7 gene regions with primers (blue arrows), analysis via capillary electrophoresis (like Figure 1), and comparison of the sequences to known sequence types (STs). PFGE workflow (B) involves enzymatic cutting of the bacterial chromosome followed by gel electrophoresis that alternates pulses of current in various directions to separate DNA fragments at higher resolution. The band patterns are compared between isolates to determine relatedness. NGS workflow (C) involves preparation of libraries from fragmented chromosomal DNA. Reads are assembled (either aligned or de novo as in Figure 1) to form contiguous sequences (contigs). From these contigs, thousands of genes or alleles are compared for similarity between isolates. An allelic distance, or number of alleles that are different between isolates, is calculated, and this can be visualized as the bubble and line plot shown above.
The problem with the methods above is that they are all limited in resolution. For viruses, the gene segment(s) may appear 100% identical between samples, but in reality, the viruses could be unrelated and display unrealized variation in a different gene region.8⇓-10 Conversely, PFGE patterns could be falsely different due to a single-nucleotide variation (SNV) that results in new restriction sites and thus different banding patterns.11 Furthermore, these methods are highly specialized for a given bacterial species or virus, requiring development and maintenance of a large number of protocols to capture all clinically relevant viruses and bacteria. The recent introduction of next-generation sequencing (NGS) technologies may allow for improvements over these conventional and historical methods because of higher resolution, sensitivity, specificity, and a “one-size-fits-all” workflow. The reasons for these possible improvements will be discussed in detail throughout this review. This review will also highlight applications of NGS to outbreaks with focus on methods (both wet bench and bioinformatics) and important related workforce needs.
HOW DOES NGS WORK FOR WHOLE VIRAL GENOME SEQUENCING?
An introduction to NGS technologies and metagenomics is provided in the companion introductory review, and detailed reviews have been published.12,13 Briefly, shotgun metagenomic sequencing of all RNA and/or DNA present in a clinical specimen can allow one to unbiasedly interrogate a sample for the presence of any viral sequence, avoiding selection bias. Reads of viral sequence can then be assembled to create a full-length viral genome for downstream comparisons (Figure 1B). This can be done via de novo assembly, whereby reads are strung together where they overlap (Figure 1B) to generate a consensus sequence. Some de novo assembly pipelines require knowledge of programming language, though commercially available software options with graphical user interfaces (GUIs) do exist. De novo assembly approaches also have specific computer processor and random-access memory requirements that must be considered prior to adoption.
Alternatively, reads can be aligned or mapped to a viral genome reference sequence (Figure 1B) using GUI software with greater speed and fewer hardware requirements. The downside to using reference sequences for mapping is that sequences that are not a close enough match may not map to the reference viral genome. This can lead to gaps in sequence, particularly if the virus is recombined (ie, fusion of 2 different strains of the virus). Thus, selection of the method to generate a whole viral genome may impact results.
The sensitivity of NGS for the detection of viral sequence directly relates to the viral load (covered in a companion review in this Focus series)14 but also relates to the depth of sequencing. In other words, to obtain coverage of the entire viral genome, one may simply need to sequence more reads of the library. Many groups have shown there is an inverse linear correlation between the crossing cycle threshold of a viral polymerase chain reaction (PCR) and reads for the same virus via shotgun metagenomic NGS (mNGS) when normalized to the total reads for that sample.15⇓⇓⇓⇓-20 However, as shown in Figure 3, the relationship is not always perfectly linear for each sample. In a published investigation into an adenoviral outbreak, we assessed the reads of adenovirus normalized to the total number of NGS reads (to account for variability with library pooling) and compared that with the crossing cycle threshold for the same samples via real-time PCR.9 As shown in Figure 3, there are samples that share the same crossing cycle threshold value but have more than 10-fold variation in the number of normalized reads for adenovirus. Not surprisingly, samples with lower background host/bacterial nucleic acids (eye and environmental samples) had significantly higher reads of adenovirus at the same Ct value compared with samples with higher background host/bacterial nucleic acids (respiratory samples). Taken together, these data mean that one cannot necessarily predict whether assembly of a whole viral genome will be possible at the outset, and repeat, deeper sequencing may be required, especially in samples with high host/bacterial background.

Normalized adenoviral reads are higher for samples with less background nucleic acid when adjusted for viral titer. The percent of adenoviral reads normalized to the total number of NGS reads is displayed on the x-axis. The adenovirus crossing cycle threshold (surrogate for viral titer) by real-time PCR is displayed on the y-axis. Samples with lower background human nucleic acid are in red and blue (eye and environmental samples, respectively), whereas samples with high human background are in green (respiratory samples). Mean CT -adjusted percent AdV per total reads was 5.87 times higher for eye specimens compared with respiratory specimens and was significantly different by ANOCOVA analysis (p = 0.017). CT, real-time PCR crossing cycle threshold.
Other factors that are important after assembly but prior to analysis include the depth of coverage for the whole viral genome. There is currently no set standard for minimum depth of coverage overall or at each nucleotide position. In general, the greater the depth of sequencing, the more confidence one has for calling an SNV or insertion/deletion (indel) real. For human genomics, there is also no established standard to call a single nucleotide variant in whole exome sequencing, so clinical genomics laboratories must validate their own criteria.21 Parallels can easily be drawn to viral sequence analysis.
There are enrichment strategies to possibly overcome samples with low viral titers that cannot generate full viral genome sequence or sequence with low depth of coverage. These include amplification using whole viral genome-spanning large primer sets.10,22 In addition, probe sets targeting either many viruses23⇓⇓-26 or the virus of interest27 can help pull sequences out, as is currently done for clinical human exome sequencing and targeted human gene panels in human genomics laboratories. This results in several hundred- to thousand-fold enrichment of the target viral sequence25,26 and thus more coverage of the viral genome. However, these methods are highly customized and take away from the “one-size-fits-all” approach of mNGS. Furthermore, they are quite costly and time consuming.
Once the complete or whole viral genome has been assembled and the coverage is of sufficient depth, the resulting consensus sequences can be used for phylogenetic analyses. An in-depth analysis of the different approaches to strain comparison is beyond the scope of this review, but readers are referred to the following reference for a detailed explanation.13 In general, phylogenetic trees are created to assess sequence similarities and estimate possible evolutionary relationships. There are several commercially available and even free software programs to perform this kind of phylogenetic analysis28,29 from consensus sequences.
CLINICAL APPLICATIONS OF NGS FOR VIRAL OUTBREAK INVESTIGATIONS
Although these approaches are still in early stages, several groups have published examples of the utility of mNGS for viral outbreak investigation. Greninger et al was one of the first groups to describe real-time use of whole viral genome mNGS data to guide infection control interventions. In a report of suspected hospital transmission of parainfluenza 3, they were able to show 100% sequence identity between 2 patients who were epidemiologically linked via a symptomatic healthcare worker.30 The same group went on to apply real-time mNGS to an outbreak of norovirus.31 In their study, Casto et al found that there were actually 3 simultaneous genetically distinct noroviral outbreaks occurring in their institution rather than a single outbreak, as suspected via epidemiologic investigation. In a similar study, Brown et al retrospectively performed mNGS on 182 norovirus-positive patient samples from the prior 2 years at their institution.32 They found 44 transmission events via whole viral genome sequencing that were not detected by conventional infection control investigations. These studies highlight the value of mNGS for outbreak investigation, as transmission dynamics can be far more complex than appreciated via conventional epidemiologic methods. Finally, mNGS can be useful for linking the suspected source of transmission via environmental sampling. In an investigation of a neonatal intensive care unit adenoviral outbreak, Sammons et al9 were able to demonstrate 100% identity across the entire adenoviral genome for outbreak cases and samples taken from ophthalmologic equipment. Although the ophthalmologic equipment was already determined to be PCR positive for adenovirus and thus the likely source of transmission, the whole viral genome sequence data helped justify significant procedure changes for ophthalmologic equipment use and cleaning.
HOW DOES NGS WORK FOR WHOLE BACTERIAL GENOME SEQUENCING?
Unlike virus culture, bacterial culture is still routinely used as standard of care in clinical microbiology laboratories. This means that isolates are readily available in most cases for sequencing. We will discuss some notable exceptions later in the review. DNA can be extracted from colonies on agar plates and libraries prepared in a similar workflow to metagenomics (Figure 2C). The ability to sequence the isolated colony results in ~100% of sequence reads belonging to the isolate of interest rather than wasted human and colonizing flora reads. The other major difference between viral sequencing and bacterial sequencing is the genome size. Clinically relevant viruses range from under 10 000 base pairs (eg, rhinovirus, enterovirus, norovirus) to the order of 100 000 base pairs (herpesviruses), whereas bacterial genomes are several million base pairs. This makes bacterial genomes computationally more challenging to work with. The other major hurdle to bacterial whole-genome sequence analysis relates to sequencing technology. The majority of clinical and research laboratories use short-read sequencing platforms (eg, Illumina, Ion). Instruments capable of sequencing longer reads are either not cost effective for most clinical laboratories (eg, PacBio) and/or are still under investigation due to higher error rates (eg, Nanopore). The use of short-read sequencers works well for assembly of clinically relevant viral genomes because of their smaller size and lack of repetitive gene regions. Conversely, for bacterial whole genomes, short reads lead to gaps in sequence assembly due to high numbers of repetitive gene regions (Figure 2C).12 Thus, complete bacterial genome assembly is typically not possible with short-read sequencing. Instead, a mostly complete genome can be assembled and further analyzed. As with viral sequencing, there are no set standards for depth of coverage for bacterial whole-genome sequencing. In general, most groups publish targeting around 100-fold coverage across the bacterial genome.33 However, laboratories must validate their own criteria for acceptance.
One approach to analysis of the incomplete genome is termed core genome MLST (cgMLST).34 Unlike conventional MLST via Sanger sequencing, which only looks at ~7 gene regions, cgMLST schemes interrogate thousands of gene regions, or alleles. cgMLST schemes are published for most clinically significant bacteria, and the list of alleles is species specific. Depending on the analysis approach, more than 97%–99% of these genes/alleles must be present and shared between bacterial species. The allelic pattern (ie, the identity of the sequence at each allele) is compared across isolates, and an allelic distance number is calculated (ie, the number of alleles that are different between 2 isolates). This number is used to define relatedness of the isolates. There are published cutoffs for many species, but, as with depth of coverage, there is variability from lab to lab. For example, in one publication applying a commercially available software for cgMLST analysis, a cutoff of ≤8 allelic differences should define relatedness of methicillin-resistant Staphylococcus aureus (MRSA) isolates in their population.35 In other studies, the allelic cutoff was larger, ranging from 18–24 allelic differences for related MRSA isolates.36 Thus, laboratories may need to validate their own cutoffs for species relatedness via cgMLST.
Several commercially available or web-based pipelines with GUI are capable of performing cgMLST analyses. These include Bionumerics (Applied Maths, Sint-Martens-Latem, Belgium) and Ridom SeqSphere+ (Ridom, Munster, Germany) software programs that can be purchased as well as Pathogenwatch, Center for Genomic Epidemiology, and Enterobase, which are free web-based programs. Some require previously assembled contiguous sequences (contigs) as the input for analysis rather than the raw sequencing fastq files. There are also commercially available and free web-based programs with GUI for this assembly step, including Sequencher (Gene Codes Corporation), Geneious (Biomatters Ltd.), CLC Genomics workbench (Qiagen), and Patric.
Other schemes beyond cgMLST are published for bacterial whole-genome sequence data, and the other most common approach is SNV analysis. Unlike cgMLST, these methods require bioinformatics resources that are generally beyond the scope of most clinical microbiology laboratories.37,38 Furthermore, they are challenging to reproduce between laboratories because of lack of standardized analysis criteria. Specifically, the reference strain used for SNV calling is highly variable between laboratories, thus making lab-to-lab comparisons of data impractical. It is important to note that each method, including cgMLST, has a source of bias and associated limitations.
CLINICAL APPLICATIONS OF NGS FOR BACTERIAL OUTBREAK INVESTIGATIONS
The first description of a cgMLST-type application for real-time bacterial outbreak investigation was performed in 2011 during the large multinational outbreak of Shiga toxin–producing Escherichia coli type O104:H4.39 The study by Mellman et al identified the strain as coming from an enteroaggregative E. coli backbone with acquisition of enterohemorrhagic E. coli genes, including Shiga toxin and extended spectrum beta-lactamase genes. These data were key in dispelling hypotheses that the strain may have been artificially introduced as a bioterrorism event. In another prospective application, the same group applied cgMLST analyses to isolates from multidrug-resistant organism surveillance during 2 different time periods.33 This included MRSA, vancomycin-resistant Enterococcus, and multidrug-resistant E. coli and Pseudomonas aeruginosa during a period of either (1) active patient isolation based on screen-positive results or (2) no isolation regardless of screen results. The authors found that there was no increased transmission of strains between patients during the second period (no isolation regardless of screen results). They estimated 200 000 Euros in savings for avoiding isolation during period 2 compared to period 1. These data support the use of whole bacterial genome sequencing for improved resolution of transmission events and potential cost-benefits for hospitals employing these protocols.
OTHER CONSIDERATIONS FOR BACTERIAL WGS
One major limitation to widespread clinical application of cgMLST or other bacterial WGS approaches is the lack of bacterial isolation as many laboratories move toward multiplex molecular panels in place of culture. For example, gastrointestinal bacteria responsible for community-acquired diarrhea are rarely cultured in clinical laboratories now because of the adoption of faster, Food and Drug Administration–cleared panels that have superior sensitivity. As a result, protocols need to be developed to assemble mostly whole-genome sequences from metagenomic datasets directly from stool. This is obviously challenging when one considers the diversity of bacteria present in a matrix, such as stool, and the contribution of plasmids that are not part of the chromosome and are easily transmitted between species in a community. In one study applying mNGS to stool from patients identified as part of the European O104:H4 outbreak, only 67% of culture-positive specimens had genome sequence from the responsible strain identifiable.40 Deeper sequencing may enhance detection; however, in the publication above, the authors knew the sequence of the pathogen they were looking for. What would happen in cases wherein the pathogen was unknown and mNGS was employed to investigate an outbreak remains to be explored? Similar protocols are being explored for Mycobacterium tuberculosis and Clostridioides difficile whole genome outbreak investigation via mNGS,41,42 as these pathogens of high infection control significance are either difficult to culture or not routinely cultured, respectively, in the clinical microbiology laboratory.
WORKFORCE NEEDS FOR NGS IN OUTBREAK INVESTIGATION
As with other applications of NGS described in this series, proficiency in wet-bench methods and some degree of bioinformatics experience are required for adoption of NGS for outbreak investigation. As the questions are distinctly different from mNGS for pathogen diagnosis and NGS for antimicrobial resistance determination, so too are the workflows and analyses, adding further complexity to the clinical microbiology laboratory. Before creating and validating protocols, clinical laboratory scientists must consider the range of pathogens that could lead to an outbreak as well as the sample types that may be implicated. As mentioned above, the depth of sequencing required will vary by pathogen load (inferred from crossing cycle threshold via real-time PCR) and specimen type. Clinical laboratory scientists should understand how to weigh these variables when pooling libraries. Furthermore, as RNA and DNA viruses have both been described in hospital outbreaks, protocols for both types of mNGS are necessary. Finally, analysis requires understanding of the differences between viral and bacterial genome assembly. Although it is likely that incorporation of individuals with formal training (eg, at least a master’s degree) in bioinformatics into the clinical microbiology laboratory will be a necessity in the future, it is important to expand the skillset of clinical laboratory scientists to include some basics on NGS analysis. Formal workshops are available for some of the programs previously mentioned, including sessions at scientific conferences. In addition, companies can provide on-site training, but both of these options come at added cost. Tutorials on whole-genome sequence analysis are provided for free by analysis software programs, with short-term free trials available prior to software purchase.
Finally, institutional support is critical for successful implementation. This includes financial support for reagents, equipment, and clinical laboratory scientist time, as these protocols, particularly the RNA library preparation, are quite time consuming. Since these are not billable tests, laboratories may have to partner with infection control programs to make a financial case for the benefits of outbreak investigation via NGS. More studies like references33 showing the potential cost benefits of NGS approaches are needed.
SUMMARY
Whole-genome sequencing will no doubt replace conventional methods, including PFGE and Sanger sequencing, for outbreak investigation. As NGS becomes more common for diagnostic applications in the clinical microbiology laboratory, it will be easier to combine outbreak sample or isolate libraries batched in clinical runs to achieve cost-effective sequencing. Appropriate training of clinical laboratory scientists in NGS methods and at least basic bioinformatic analysis concepts will become critical for rapid-outbreak investigation.
- Received June 1, 2019.
- Accepted September 5, 2019.
American Society for Clinical Laboratory Science
References




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- LEARNING OBJECTIVES
- ABSTRACT
- INTRODUCTION
- HOW DOES NGS WORK FOR WHOLE VIRAL GENOME SEQUENCING?
- CLINICAL APPLICATIONS OF NGS FOR VIRAL OUTBREAK INVESTIGATIONS
- HOW DOES NGS WORK FOR WHOLE BACTERIAL GENOME SEQUENCING?
- CLINICAL APPLICATIONS OF NGS FOR BACTERIAL OUTBREAK INVESTIGATIONS
- OTHER CONSIDERATIONS FOR BACTERIAL WGS
- WORKFORCE NEEDS FOR NGS IN OUTBREAK INVESTIGATION
- SUMMARY
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- cgMLST - core genome MLST
- GUI - graphical user interface
- MLST - multilocus sequence typing
- mNGS - metagenomic NGS
- MRSA - methicillin-resistant Staphylococcus aureus
- NGS - next-generation sequencing
- PCR - polymerase chain reaction
- PFGE - pulsed field gel electrophoresis
- SNV - single-nucleotide variation
- ST - sequence type
- WGS - whole-genome sequencing
- whole-genome sequencing
- outbreak investigation
- next-generation sequencing
- metagenomics