


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Address for Correspondence: Robert J. Tibbetts
, Henry Ford Health System, rtibbet1{at}hfhs.org
LEARNING OBJECTIVES
1. Explain the importance of antimicrobial susceptibility testing in management of patient care.
2. Describe various methods of antimicrobial susceptibility testing and their limitations.
3. Describe the governance and development of minimum inhibitory concentration breakpoints and challenges associated with global standardization of antimicrobial susceptibility testing and interpretation.
ABSTRACT
wAntimicrobial susceptibility testing (AST) results are among the most important pieces of information a clinical microbiology lab can release, and clinicians rely heavily on them to care for their patients. Because of the clinical importance of these results, it is imperative that testing is performed under optimal conditions with standardized approaches to quality control, interpretation, and reporting. There are a variety of in vitro methods to determine bacterial antimicrobial susceptibilities as well as interpretive criteria and testing limitations that have been vetted through the Clinical and Laboratory Standards Institute and the European Committee on Antimicrobial Susceptibility Testing. The goal in Part I of this two-part section is to review the general background, breakpoint development, and governance of AST testing, and the goal in Part II is to review the standardization of the various methods of AST, their limitations and associated challenges, and the future of AST using newer technologies.
- AST - antimicrobial susceptibility testing
- AUC - area under the curve
- BMD - broth microdilution
- CFU - colony-forming unit
- CLSI - Clinical and Laboratory Standards Institute
- CSF - cerebral spinal fluid
- ECV - epidemiologic cut-off value
- EMA - European Medicines Agency
- EUCAST - European Committee on Antimicrobial Susceptibility Testing
- fCmax - peak free drug concentration
- FDA - Food and Drug Administration
- fT - free drug concentration
- I - intermediate
- MIC - minimum inhibitory concentration
- NS - nonsusceptible
- NWT - non–wild type
- PAE - post-antimicrobial effect
- PCR - polymerase chain reaction
- PD - pharmacodynamics
- PK - pharmacokinetics
- R - resistant
- S - susceptible
- SDD - susceptible dose-dependent
- USCAST - National Antimicrobial Susceptibility Testing Committee for the United States
- UTI - urinary tract infection
- WT - wild type
PART I. INTRODUCTION
Clinicians rely greatly on all patient results generated from the microbiology laboratory; however, it has been suggested that antimicrobial susceptibility testing (AST) results may be the single most important to aid in the treatment of their most critically ill patients.1⇓-3 Not only do AST results drive infection-specific management, temporally collected data can be used to drive empiric antimicrobial therapy in the form of yearly antibiograms and can be used to monitor the development and spread of resistance mechanisms.2,4 The use of dilution-based AST to gauge antimicrobial activity has been around since the discovery of penicillin in 1929, and Alexander Fleming described a tube-based dilution method determining penicillin minimum inhibitory concentrations (MICs) of fungal culture filtrates.5 In its infancy, the performance of AST varied significantly from lab to lab regarding the entire process (ie, composition of media, inoculum size, incubation conditions, and antimicrobial purity).4 However, over the past 30 years, AST has undergone a significant degree of procedural standardization and oversight by various agencies [ie, Clinical and Laboratory Standards Institute (CLSI) resulting in very robust, reproducible, and interpretable results from lab to lab across the world]. Although there are a number of in vitro methods to determine the potential in vivo efficacy of a particular antimicrobial agent, not all are the same, and discrepancies have been reported.6 The general lack of new antimicrobial drug classes and the high level of resistance to newer classes are a major cause of concern and demand robust testing and interpretive criteria. Therefore, the goal of the following review is to outline these tests and their limitations, governance and development of interpretive criteria, current challenges, and the future direction of AST.
GOVERNANCE OF ANTIMICROBIAL SUSCEPTIBILITY BREAKPOINT DEVELOPMENT AND TESTING MICS
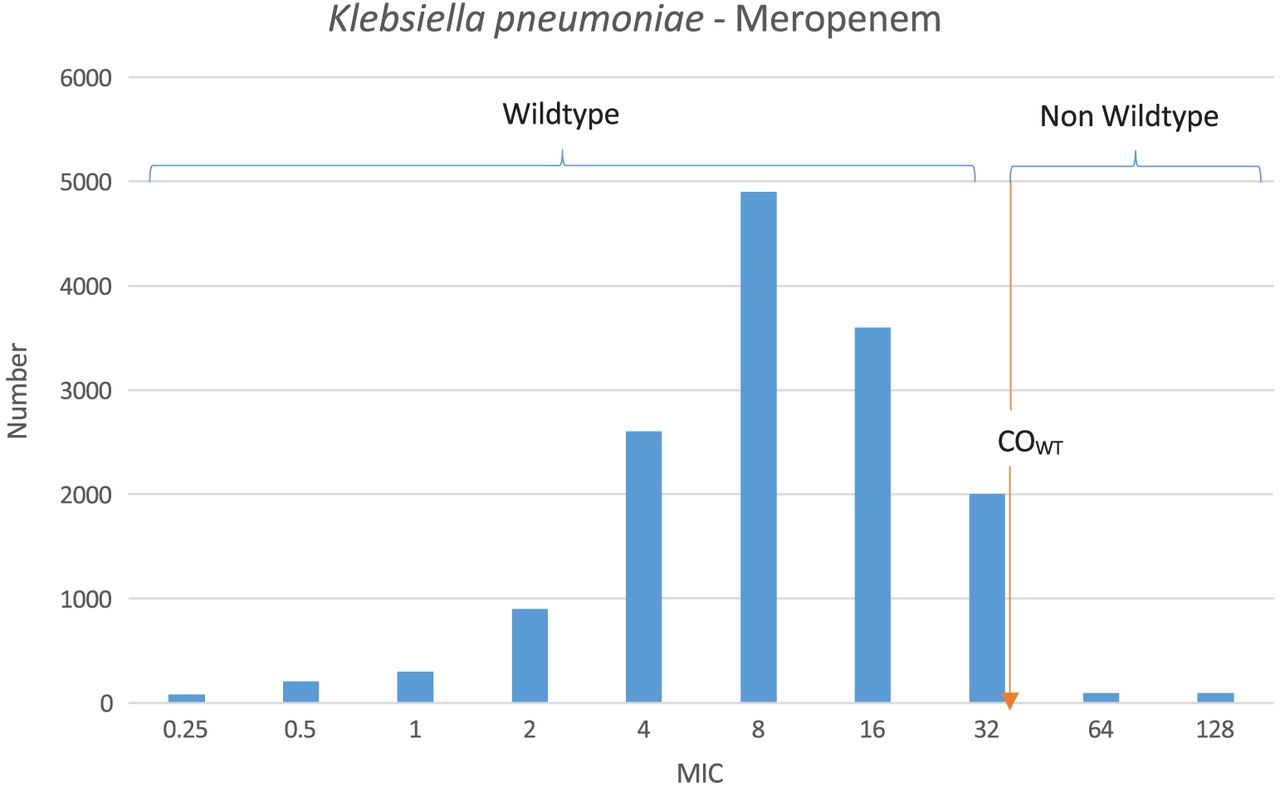
In AST, the MIC is a numeric value that indicates the lowest concentration of an antimicrobial agent that inhibits growth of a bacterium in vitro.1 The ranges of MICs are fundamental in the formation of susceptibility testing methods; they allow for comparison against achievable human serum levels for determining susceptibility and therefore provide the foundation for categorizing the numeric MIC results of AST to predict clinical outcome.2,7 According to the CLSI, there are six breakpoint categories that provide clinicians the information to drive therapeutic decision making.2 Traditionally, these were defined as: “susceptible” (S), indicating that when using Food and Drug Administration (FDA)-approved standard dosing the antimicrobial concentrations are typically achievable for clinical cure; “intermediate” (I), suggesting that the bacteria have MICs at or near the high end of achievable antimicrobial concentrations and response rates may be lower than susceptible isolates; and “resistant” (R), indicating that achievable antimicrobial concentrations are not likely to have a favorable clinical response.1 It is important to note that most breakpoints are based on achievable levels of a particular antimicrobial in serum only; therefore, depending on where the infection is located, these breakpoints may not necessarily correlate with clinical resolution using the FDA-approved standard dose and duration of treatment.8 More recently, two additional breakpoint designations have been developed: “susceptible dose-dependent” (SDD) (similar to, yet has replaced, “intermediate” for two drugs; suggests that with altered dosing strategies, antimicrobial concentrations will be achievable for clinical cure) and “nonsusceptible” (NS) (used when only a susceptible designation has been made because of the absence or rarity of bacteria resistant to that antimicrobial).1,8 Most recently a sixth designation is the epidemiologic cut-off value (ECV).9 ECVs are based solely on the in vitro MIC distribution of a particular antimicrobial and bacterium and not on in vivo or clinical cure data; therefore, they are very different than true MIC breakpoints.1,9 The ECV is determined using either a computer algorithm or, oftentimes, simply based on the wild-type (WT) distribution of the drug/bug combination, and any MIC that is greater than this ECV is considered to be a non-WT (NWT) phenotype as described below and in Figure 1.1 The benefit of using an ECV is that the ECV value is often low, so it is more sensitive to detecting small changes in antimicrobial susceptibility and new/emerging mechanisms of resistance.10 Since the clinical relevance of ECVs has not been determined, it is very important to note that ECVs and breakpoints are not interchangeable but rather are used primarily to monitor the emergence of NWT strains of a particular bacterium.1,10
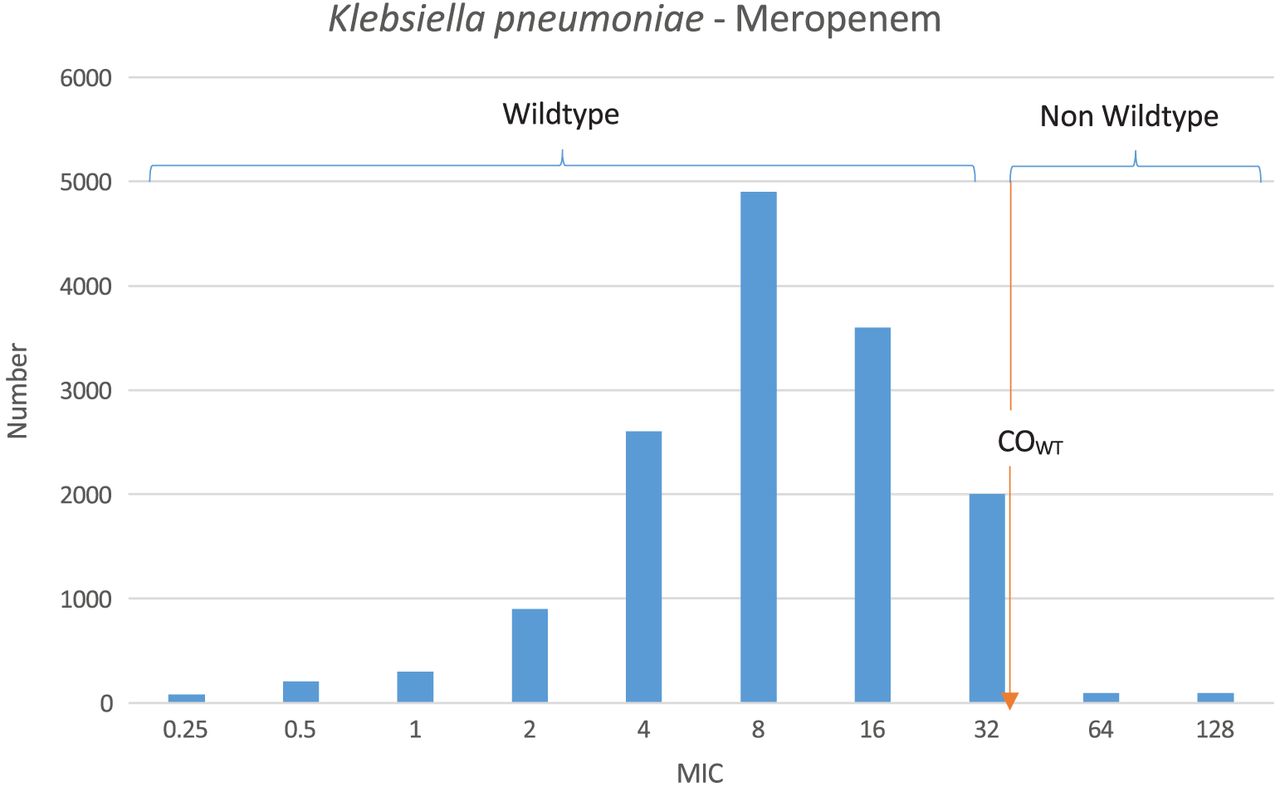
MIC distributions for a single bacterium/antimicrobial pair. As shown, the wild-type population appears to be log-normally distributed at the lower MICs. COWT (ie, ECV) is the calculated wild-type cutoff value and forms the basis of the “susceptible” breakpoint. MICs greater than this are considered non–wild type.
The conventional method to determine the MIC is by using serial two-fold dilutions of a particular antimicrobial agent using either an agar dilution or broth microdilution (BMD) technique.7 This dilution was initially chosen for the ease of making the dilution series; however, the choice turned out to be a serendipitous, in part because in the absence of a resistant determinant, any given bacterial species will have an MIC range to any given drug in a statistically normal distribution.7 This distribution then allows investigative organizations to determine the WT vs NWT MICs used, in part, to determine the categorical breakpoints.7,8 In very early evaluations of BMD techniques, it was determined that 90% to 95% of MIC results were within ±1 dilution from the median for most drug/bug combinations.11
A downside to this doubling dilution is that any error in a doubling dilution result (eg, between two different methods or within the normal “error rate,” it will represent at least a 2-fold difference).4 To be exact, when the lab reports that an organism has an MIC of 2 ug/ml, what they really mean is that it has an MIC of between 1 to 4 ug/ml.12 Furthermore, if this happens to occur at an established breakpoint, it may result in a different categorical designation, which may have unintended clinical consequences.4 Two very important scenarios related to this allowable error rate, whether it is between two different methods of AST or multiple AST results from a single method, are major errors and very major errors.9,13 The former is when the reference result is susceptible and the other result is resistant, and the latter is when the reference result is resistant and the other is susceptible. For a more thorough review of these errors and the allowable rates for AST testing, see the CLSI M52 and M23 documents.14,15 Additionally, in a large Phase 3 clinical trial consisting of ~1500 patients with a gram-negative infection treated with cefotaxime, it was noted that 64% of those patients with a cefotaxime resistance bacteria and 93% with a susceptible bacteria showed a favorable clinical outcome.12 These results have been corroborated in additional studies,16,17 and based on this and several other studies, Rex and Pfaller coined the phrase “the 60/90 rule”; that is, infections due to susceptible isolates respond favorably to treatment ~90% of the time, and those with infections due to resistant isolates respond favorably ~60% of the time, although this rule generally does not apply to immunocompromised patients and/or those with multiple comorbidities.18
Lastly, breakpoint utilization is described in the form of disc diffusion AST (see more in the following). Breakpoints for the determination of S, I, or R are based the “zone of inhibition” of growth in millimeters around a paper disc impregnated with a fixed concentration of antimicrobial agent.9 Once the actual MIC breakpoints have been established, the zone diameter breakpoints can then be determined. The primary method to do this is to test the bacteria and antimicrobial agent using both the broth dilution and disc diffusion methods and creating a scatter plot of the results with zone diameter on the y-axis and MIC on the x-axis.7,8 Visual inspection of the correlation of MIC vs the zone diameter has allowed for the determination of where the breakpoints should be in millimeters (Figure 2).7,8 For a much more detailed review of both methods of breakpoint development, please see Turnidge and Paterson.8

Comparison of zone diameters with MICs of a hypothetical antimicrobial (reprinted from reference7 with permission from the publisher). For this type of study, both the MICs and zones of inhibition are determined for a series of bacterial isolates. These values are then plotted in graphical form. The numbers within the boxes refer to the number of isolates with a particular MIC (y-axis) that matches a particular zone of inhibition (x-axis). For example, there were 12 isolates with an MIC of 0.12 ug/ml that also had a 40-mm zone of inhibition. Isolates that had MICs that were in the resistant range but were susceptible by the zone of inhibition are considered to be “very major errors.” MICs that were in the susceptible range but which the zones of inhibition indicated as resistant are considered to be “major errors.” When the MICs were in the intermediate range but their zone of inhibition was either susceptible or resistant, they are considered to be “minor errors.”
GOVERNANCE
In the United States, there are three organizations involved in AST development and breakpoint interpretation guidelines: the FDA Center for Drug Evaluation and Research, the CLSI, and the National Antimicrobial Susceptibility Testing Committee for the United States (USCAST).2,10,19 The CLSI is an international, interdisciplinary, not-for-profit, standards-developing educational organization whose goal is to promote the development and use of consensus-developed testing and interpretive guidelines for use in the health care industry.1 Similarly, USCAST describes itself as “part of a global network dedicated to fighting the rise of antibiotic resistance through scientific, educational, and policy-creating activities.”20 In contrast, the FDA is a government agency within the Departments of Health and Human services whose primary function, in part, is in the regulatory oversight of in vitro medical devices, for example, AST methodology. In Europe, the European Committee on Antimicrobial Susceptibility Testing (EUCAST) and European Medicines Agency (EMA) function in the same capacity, respectively, with the primary difference being that EMA funds EUCAST, whereas the FDA does not fund CLSI.10 Although their goals are the same—to develop functional breakpoints—how they go about analyzing the results can vary, and as a consequence, the breakpoints do not always match among them.2,10
BREAKPOINT DEVELOPMENT
The process of breakpoint development is complex and involves numerous sources of data for the initial setting and continued data collection once approved and implemented. There are four main processes: 1) MIC distribution and WT cutoffs, 2) pharmacokinetc (PK)/pharmacodynamic (PD) data from both animal models and human studies, 3) clinical and bacteriological outcome data from clinical studies, and 4) genotypic and phenotypic in vitro resistance markers (described in Part II. Methods of AST in the clinical lab and individual limitations).8 In vitro modeling (ie, “Monte Carlo simulation”) is also used to predict breakpoints.21 Turnidge and Paterson produced an excellent review of Monte Carlo simulation.8
The first step in the breakpoint development process is to collect MIC distribution data by testing thousands of individual bacterial/antimicrobial combinations using either BMD or agar- dilution methods.8 These data are arranged into histograms wherein the obvious WT/NWT cutoffs can be observed, forming the basis of the susceptible breakpoint (Figure 1).8 If there is a single normal distribution, it would suggest only a susceptible population exists, also known as the WT phenotype21; however, a bimodal normal distribution would suggest that the bacterial population includes resistant isolates, also known as the NWT phenotype.8,21
The next step is an analysis of PK/PD data. PK is defined as the activity of the drug over time in the patient at the site of infection, including absorption, distribution, and elimination, whereas PD is the effect of the drug on the infection and disease, including pharmacological and toxicological.10,22 Taken together, PK/PD is the study of the relationship between the activity of the drug and bactericidal/bacteriostatic effects in vitro and, therefore, by extension, clinical outcomes.8 The four indices incorporated by a drug’s PK/PD and the MIC used to determine the killing effect of a particular antimicrobial are the length of time the free drug concentration (fT) remains above the MIC (fT > MIC), the ratio of peak free drug concentration (fCmax) to the MIC (fCmax:MIC), the ratio of the area under the curve (AUC) of a 24-hour time curve of drug concentration to the MIC (AUC:MIC), and the post-antimicrobial effect (PAE) is the duration of bacterial regrowth following removal of the antimicrobial.12 Using these indices, antimicrobials can be classified into three types: 1) those with time-dependent action with no or short PAE, 2) those with time-dependent action and long-lasting PAE, and 3) those with prominent concentration-dependent actions (Table 1).7 These indices allow for the determination of the maximum breakpoint (greater than this value would indicate resistant) that would allow the achievement of optimal antimicrobial efficacy using approved dosing schedules for the antimicrobial in question.7
Various indices of several families of antimicrobial agents based on the type of antimicrobial and post-antimicrobial effect.
It is important to note that PK/PD breakpoints are established using data from plasma/serum concentrations, which do not necessarily predict success in tissues or other body sites, such as the cerebral spinal fluid (CSF) or synovial fluids.8 For example, an antimicrobial that tests susceptible in vitro on a bacterium from CSF may be a false susceptible if the antimicrobial does not cross the blood brain barrier.1 Conversely, false resistance occurs very often on urinary isolates, as some drugs used for UTIs concentrate in much higher levels than can be achieved in serum. For a very detailed review of PK/PD including in vitro and in vivo animal modeling, see Turnidge and Paterson.8
Once the MIC distribution and the WT/NWT breakpoint have been determined (plus Monte Carlo simulation, as applicable) and PK/PD parameters have been calculated, then tentative breakpoints can be determined and the study can proceed with clinical trials.9 Clinical trials are designed to test these tentative breakpoints by measuring the bacterial response rate or successful eradication of the bacteria from the site of infection as well as the clinical response rate.9 In the United States, the CLSI and the FDA are the primary stakeholders in breakpoint development, and both use some or all of the aforementioned data sets to do so; however, the breakpoints can and often do differ.1,7,8 The clinical implications for these differences can be significant since FDA breakpoints are usually set when a new drug is introduced; some of the current FDA breakpoints were set decades ago prior to known resistance mechanisms and may be too high or too low, resulting in inappropriate antimicrobial usage.10 In addition, pharmaceutical companies that are granted FDA approval for an antimicrobial can only use the FDA-approved breakpoints in their product literature, and as such, clinical labs must use these breakpoints despite the availability of more current or revised CLSI breakpoints.10
Taken together, these discrepancies can and do create confusion for clinical microbiologists, AST device manufacturers, and clinicians.10 Therefore, the question of whether the CLSI and the FDA will band together to agree upon a single set of breakpoints has been posed numerous times.21 Despite the two organizations working together, this has not happened until just recently. In December of 2016, the “21st Century Cures Act” was signed into law with the goal to “ensure a more efficient process by which to update susceptibility test interpretive criteria to recognize antimicrobial resistance.”19 The first action from this was that the FDA created an Antimicrobial Susceptibility Test Interpretive Criteria website that recognized and accepted breakpoints that were developed by the CLSI, which were typically published separately in the CLSI M100 and other CLSI documents, as well as their own breakpoints.19 Although there are challenges ahead, this is a great first step in the unification of breakpoint development between the three organizations involved in the United States to develop standardized MIC interpretation and categorization in order to predict clinical cure with both high sensitivity and specificity.
PART II. METHODS OF AST IN THE CLINICAL LAB AND INDIVIDUAL LIMITATIONS
Despite the potential differences in breakpoints, there is a global consensus on the way AST should be performed and the methods to perform them. Since the inception of AST by Fleming in 1929 using a tube-broth method, additional methods quickly arose out of necessity; ease of use, standardization, consistency, and miniaturization were the driving factors. Antimicrobial activity can now be determined using a wide variety of different in vitro methods. These methods include disk diffusion, microbroth dilution, and Etests, which are manual or instrument-based automated methods.12 With the exception of the disk diffusion method, all others provide actual MICs, whereas disc diffusion results in categorical results (S, I, or R).
One of the biggest challenges in performing and resulting AST, regardless of the method used, is the clinical precision of the results and their predictive value for clinical cure.4 A number of studies have been performed that have sought to determine the major source(s) of AST variability, which determined that there are both biological and technical causes.4 Biological variability exists between different bacteria regarding growth phase/doubling times or various nutritional or temperature requirements that may alter growth, whereas technical variability arises from nonstandardized preanalytic and postanalytic handling of the bacteria and the media used during testing.4 To that end, both the CLSI and EUCAST have standardized these processes to ensure the least amount of variability, including media types, incubation times, and temperature, bacterial concentration, and assay performance, all of which are published in the CLSI M07-A11 document.3 In addition, both organizations provide recommendations on which bacteria/antimicrobial combinations to test, MIC categorical interpretation, and which bug/drug combinations to report in groups.1
TEST METHODS
Phenotypic AST can be divided into two primary categories: broth dilution and agar-based methods, with the latter being divided into agar diffusion and agar dilution.3 Broth dilution utilizes a predefined volume of a liquid growth medium, typically cation-adjusted Mueller-Hinton (MH) broth, to which a known concentration of antimicrobial is added to the first tube. Serial two-fold dilutions are then made to subsequent tubes to the desired end-point dilution.3 A known concentration of bacteria (inoculum) is made and added to each tube. For this method, the starting inoculum is 1–5 × 105 CFU/ml. The tubes are incubated at 35 °C for 18–24 hours; the first tube showing no growth of bacteria (no turbidity in the broth) is the MIC.
This process can occur in any starting volume; however, BMD is most common using 96-well disposable plates with 0.1 ml of broth.3,9 Furthermore, BMD is considered the “gold standard” of AST comparison in the clinical lab. The advantage of this method is that a single bacterium can be tested against 12 different antimicrobials with up to eight two-fold dilutions, thus generating MICs, and the results are very reproducible.9 The disadvantage is the manual nature of preparing the plates and reporting results and, in the case of commercially prepared plates, the cost and the relative inflexibility of antimicrobial choice and the length of time to get new panels with newer drugs.9 This same method is what is typically used by commercially available automated systems, only the volumes are in the 10–20 ul range. The advantage of these systems is the automation, as results are determined and can be automatically uploaded into the laboratory information system and patient medical record. The disadvantages are the same as those for broth dilution listed above.
For the agar-based AST methods, the same MH broth is used but in a solid from the addition of agar, and the inoculum for agar-based methods is diluted to 1–2 x 108 CFU/ml, also known as a McFarland 0.5.3 Incubation, temperature, and time are the same as broth dilution. Two-fold antimicrobial dilutions are then made on individual agar plates, so as an advantage of this method, bacterial inoculum are plated onto the agar using a “replica plater” apparatus, allowing for inoculation of up to 32 different bacteria per plate. In doing so, each bacterial species will be placed in the exact same location on all subsequent plates. Following incubation, the first plate on which there is no colony growth is considered the MIC.3 Two major disadvantages to this method are 1) the potential for crosscontamination of bacterial suspensions using a replica plater and 2) the manual nature of making agar plates correctly, as the depth of the agar in the plate is crucial for result consistency; as such, this method is rarely if ever used in the clinical lab.
Two other agar-based methods utilize either paper discs or plastic strips impregnated with either a fixed amount of antimicrobial as in Kirby-Bauer disc diffusion or a gradient of antimicrobial agent as used in gradient strip diffusion.3 As the antimicrobial diffuses through the agar upon placement of the disc or strip, it creates either a round zone of antimicrobial with decreasing MICs moving away from the disc or an elliptical zone of MICs with the highest at the top when using the gradient strips. Lack of growth around the disc or strip is called the “zone of inhibition.” For the disc, the diameter of the zone determines if the bacteria is S, I, or R for the strip, and the lowest point at which the growth ellipsis intersects the strip determines the MIC.3,9
Lastly, genotypic AST occurs through the use of molecular-based nucleic acid amplification techniques, such at polymerase chain reaction (PCR).23 These types of assays often only look for one specific gene associated with one type of resistance, for example, the vanA or vanB genes associated with vancomycin resistance in Enterococcus species or the mecA gene associated with methicillin resistance in Staphylococcus species. Despite only looking for a single gene, the presence of the gene may predict antimicrobial susceptibility to a wide range of antimicrobials; for example, mecA positive S. aureus is resistant to all beta-lactams and not just methicillin.23 As the need has grown, there are now a number of FDA-approved commercially available “multiplex” systems that can detect multiple resistance genes simultaneously, for example, the Cepheid GenXpert® CarbaR test that detects KPC, NDM-1, IMP, VIM, and OXA- 48 carbapenemases.24 For a nice review of these systems and their targets, please see Abbott and Fang.23 The main advantages of molecular detection, in particular the commercially available systems, lie in the ease of use and turnaround time as most of these only take 1–2 hours as opposed to 18–24 hours incubation and they can detect multiple genes simultaneously. There are several disadvantages to these assays as well. First is the cost, since many of them are considerably more expensive than traditional AST in clinical labs. Second, outside of the few genes mentioned above, not all resistance genes can predict phenotypic resistance to multiple antimicrobials; for example, there are hundreds of beta-lactamases, point mutations confirming resistance to the quinolones, efflux pump mechanisms, and outer membrane porin mutations that our current genotypic methods of detection cannot easily detect.9 For a nice review on molecular detection of antimicrobial resistance determinants, see Ledeboer and Richard.25
CHALLENGES AND FUTURE DIRECTION
Two of the biggest challenges to AST are 1) the creation and adoption of standardized breakpoints and 2) testing that is timely and comprehensive. As indicated previously, the rollout of the 21st Century Cures law has begun to tackle the issues of multiple organizations defining MIC breakpoints, thereby standardizing how the MIC results are interpreted.19 In addition, the law will remove the constraints to the commercial AST developers, allowing for a more rapid development of expanded test panels and the ability to change MICs with changing breakpoints.19 This then leads to the second challenge and the most likely direction AST will move to in the future: next-generation sequencing and microbiome analysis, also known as “Resistome” analysis. The concept of the microbiome is not new; however, it is only recently that it has been used to identify not only all resistance genes in a particular bacterium but also the total microbiome to predict treatment failure and clinical resolution.26 One downside to this type of analysis is similar to that with the other molecular-based testing methods, namely that the presence of a resistance gene may not necessarily predict for phenotypic resistance. In January of 2017, van der Helm et al published a first-of-its-kind analysis of the resistome and the clinical implications thereof.26 In it they describe a cost-efficient and rapid tool to determine and analyze the resistome that will inform antimicrobial treatment as the future of AST.
SUMMARY
Since its inception, AST results have been used for appropriate antimicrobial selection and to predict successful clinical outcomes. As such, it is imperative that laboratorians perform and interpret these tests with precision and standardization. There has been a tremendous evolution of AST methods since the first days of bacterial growth visualization 70 years ago, including standardization of bacterial inoculum, media type and incubation temperatures, and duration. Organizations, such as the CLSI, EUCAST, and FDA, have developed MIC breakpoints in order to standardize the interpretation of susceptible, intermediate/SDD, or resistant. Furthermore, although the CSLI and FDA breakpoints often differed, there has been strong interest in collaborating to standardize those as well, and the first set of agreed upon breakpoints has been recently released. However, as we move into more molecular testing, the question of whether we need breakpoints exists since knowing if the bacteria will or can become resistant based on the genes in the resistome may preclude the use of a particular antimicrobial anyway.
- Received May 18, 2018.
- Accepted May 22, 2018.
American Society for Clinical Laboratory Science
References




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- AST - antimicrobial susceptibility testing
- AUC - area under the curve
- BMD - broth microdilution
- CFU - colony-forming unit
- CLSI - Clinical and Laboratory Standards Institute
- CSF - cerebral spinal fluid
- ECV - epidemiologic cut-off value
- EMA - European Medicines Agency
- EUCAST - European Committee on Antimicrobial Susceptibility Testing
- fCmax - peak free drug concentration
- FDA - Food and Drug Administration
- fT - free drug concentration
- I - intermediate
- MIC - minimum inhibitory concentration
- NS - nonsusceptible
- NWT - non–wild type
- PAE - post-antimicrobial effect
- PCR - polymerase chain reaction
- PD - pharmacodynamics
- PK - pharmacokinetics
- R - resistant
- S - susceptible
- SDD - susceptible dose-dependent
- USCAST - National Antimicrobial Susceptibility Testing Committee for the United States
- UTI - urinary tract infection
- WT - wild type
- antimicrobial susceptibility testing
- antimicrobial resistance