


This article requires a subscription to view the full text. If you have a subscription you may use the login form below to view the article. Access to this article can also be purchased.
- Samantha Giordano
- Robert Estes
- Wei Li
- Remo George
- Tosi Gilford
- Krystle Glasgow
- Heather Hallman
- Floyd Josephat
- Ana Oliveira
- Neena Xavier
- Janelle M. Chiasera
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- University of Alabama at Birmingham
- Address for Correspondence: Janelle M. Chiasera
, University of Alabama at Birmingham, chisera{at}uab.edu
LEARNING OBJECTIVES
1. Explain structure and function of the components in striated muscle contraction.
2. Describe the isoforms of troponin and explain the functional role of each isoform in striated muscle contraction.
3. Explain how mutations in the amino acid sequence or protein structure of troponin could cause changes to muscle contraction.
4. Describe the use of troponin as a clinical biomarker for cardiac diseases.
ABSTRACT
Troponin (Tn) is a heterotrimeric protein containing 3 subunits (C, T, and I) with different molecular weights and distinctive functions. The subunits of Tn work cohesively to regulate the contraction and relaxation activities of striated muscles: troponin-C (TnC) binds calcium (Ca2+); troponin-T (TnT) interacts with tropomyosin (Tm) and anchors Tn to actin; and troponin-I (TnI) inhibits the adenosine triphosphatase (ATPase) activity of the actomyosin cross-bridge and effectively blocks the myosin-binding site on actin subunits. At a genetic level, there are 8 distinctive Tn genes (isoforms) that code for tissue-specific heart and skeletal muscle protein subunits: TNNI1, TNNI2, TNNI3, TNNT1, TNNT2, TNNT3, TNNC1, and TNNC2. The gene isoforms are regulated throughout development via posttranscriptional and posttranslational modifications. Genetic mutations in any of the 3 protein subunits could be linked to hypertrophic, dilated, and restrictive cardiomyopathies. Tn release by damaged cardiomyocytes is clinically used as a biomarker for myocardial infarction (MI), and its release into the serum is measured at specific times postinjury for diagnostic or prognostic purposes. Current tests that measure serum Tn are fifth generation assays, which have improved sensitivity and specificity compared with previous assays. However, increased serum Tn levels have been seen in chronic diseases such as Fabry disease and chronic renal disease. It is important to remember, when clinically examining a patient, that Tn levels are only one piece of the puzzle. A patient’s history or symptoms are essential for making an accurate diagnosis.
- ADP - adenosine diphosphate
- ATP - adenosine triphosphate
- ATPase - adenosine triphosphatase
- Ca2+ - calcium
- cTn - cardiac troponin
- cTnC - cardiac troponin-C
- cTnI - cardiac troponin-I
- cTnT - cardiac troponin-T
- DCM - dilated cardiomyopathy
- fsTnC - fast twitch skeletal troponin-C
- fsTnI - fast twitch skeletal troponin-I
- fsTnT - fast twitch troponin-T
- HCM - hypertrophic cardiomyopathy
- Mg2+ - magnesium ion
- MI - myocardial infarction
- mRNA - messenger RNA
- PKA - protein kinase A
- RCM - restrictive cardiomyopathy
- ssTnI - slow twitch skeletal troponin-I
- ssTnT - slow twitch skeletal troponin-T
- Tm - tropomyosin
- Tn - troponin
- TnC - troponin-C
- TnI - troponin-I
- TnT - troponin-T
INTRODUCTION
Troponin (Tn) is an intracellular protein that plays an important role in regulating striated muscle contraction.1 First discovered in 1965, it was originally named “tropomyosin-like protein”2 until 1973 when it was purified and characterized from the skeletal muscles of a rabbit as a separate complex consisting of 3 different protein subunits. Tn is a heterotrimeric protein containing 3 subunits (C, T, and I) with different molecular weights and distinctive functions.3 The subunits of Tn work cohesively to regulate the contraction and relaxation activities of striated muscles: troponin-C (TnC) binds calcium (Ca2+), troponin-T (TnT) interacts with tropomyosin (Tm) and anchors Tn to actin, and troponin-I (TnI) inhibits the adenosine triphosphatase (ATPase) activity of the actomyosin cross-bridge and effectively blocks the myosin-binding site on actin subunits. Based on the muscle type and stage of development, 3 subunits (from 8 different isoforms) come together to form various triplets in either heart or skeletal muscle (Table 1).
Troponin isoforms and chromosomal location
At a genetic level, there are 8 distinctive Tn genes (isoforms), which code for tissue- specific heart and skeletal muscle protein subunits. TnI has 3 isoforms encoded by 3 homologous genes: TNNI1 for slow skeletal muscle TnI, TNNI2 for fast skeletal muscle TnI, and TNNI3 for cardiac TnI. Similarly, TnT has 3 isoforms encoded by 3 homologous genes: TNNT1 for slow skeletal muscle TnT, TNNT3 for fast skeletal muscle TnT, and TNNT2 for cardiac TnT. TnC has 2 isoforms encoded by 2 homologous genes: TNNC1 for slow skeletal and cardiac muscle TnC and TNNC2 for fast skeletal muscle TnC. Unlike TnI and TnT, TnC does not have a unique cardiac isoform (Table 1).5
The different isoforms of Tn encode for proteins with similar but not identical function, which allow the body to fine tune muscle contraction in the cardiac, slow twitch skeletal muscle, and fast twitch skeletal muscle. The 8 subunit isoforms undergo further fine tuning at the transcriptional level via alternative splicing of the messenger RNA (mRNA). The tight regulation of Tn, specifically cardiac troponin (cTn), at both the transcriptional and the translational level play an important role in cardiac development, health, and muscle pathology.
STRIATED MUSCLE STRUCTURE
Striated muscle, which includes human skeletal and cardiac muscle tissues, is composed of individual muscle cells (myofibers) that consist of bundles of thin filamentous structures called myofibrils. The myofibrils are composed of individual functional units called sarcomeres, arranged end to end throughout the length of the myofibrils. Skeletal muscle cells are long, unbranched, and multinucleated; whereas, cardiac muscle cells are relatively short, branched, and usually contain a single nucleus. Despite these differences, both skeletal and cardiac muscles share a striated appearance because of the arrangement of proteins within the sarcomeres.
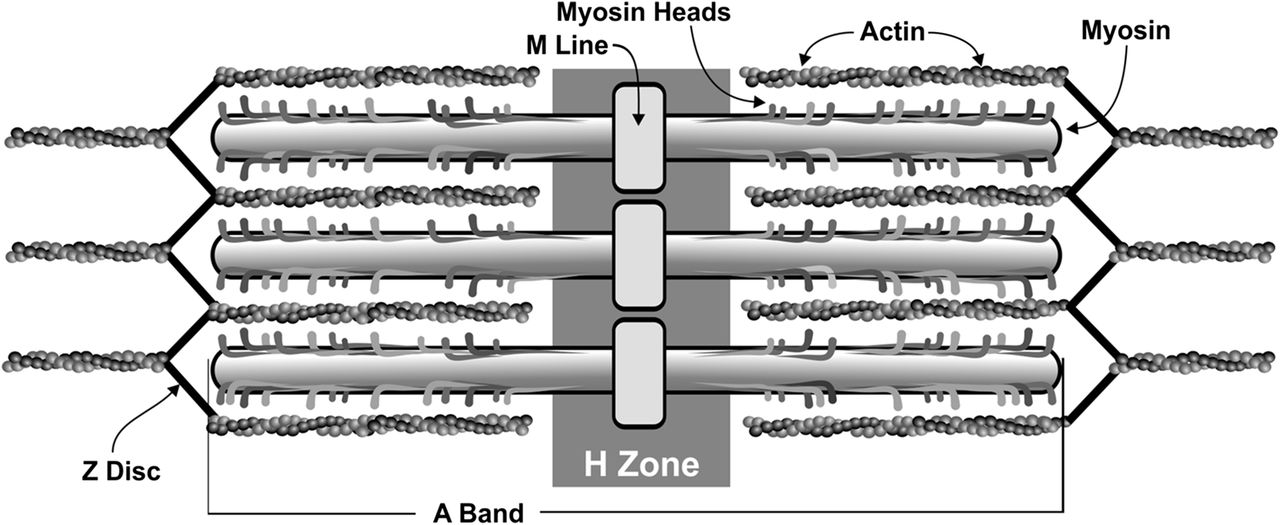
Contraction of striated muscle is a highly-regulated and coordinated process, involving interplay among multiple protein components that are arranged in specific patterns. The sarcomere is composed primarily of the proteins actin (thin filament) and myosin (thick filament). The actin is associated with Tm, which acts to block the myosin-binding sites on actin. With the troponin complex, in conjunction with Ca2+, it works to regulate the formation of cross-bridges between the thick and thin filaments. Each sarcomere runs from Z disc to Z disc, which is composed of the protein alpha-actinin and anchors the thin filaments (Figure 1). According to the sliding filament model of muscle contraction proposed in 1954, during contraction, actin and myosin interact with each other in such a way that when activated the thin filaments slide over the thick filaments, drawing the Z discs toward each other and effectively shortening the sarcomere.4,5 As successive sarcomeres shorten collectively, myofibrils shorten and contraction takes place.

Sarcomere organization. A sarcomere is the smallest functional unit of muscle contraction. It is organized from Z disc to Z disc (termed the A Band), with an M line found in the center. The functional unit consists of the actin and myosin filaments, which will slide over one another toward the M line during contraction and shrink the H zone. Image reprinted with permission of John Nagy.
TROPONIN IN MUSCLE CONTRACTION
Contraction is initiated when muscle fibers are stimulated by a nerve impulse, and cytoplasmic Ca2+ increases in skeletal muscle by release from the sarcoplasmic reticulum or in cardiac muscle by the release from the sarcoplasmic reticulum and influx of extracellular Ca2+. Tn, which is associated with actin filaments, binds the Ca2+ ions. The binding of Ca2+ to Tn subsequently displaces Tm, which exposes myosin-binding sites on actin. The 3 Tn protein subunits each play a specific role in regulating muscle contraction within the actin-myosin cross-bridge. The TnT subunit binds the whole Tn protein to Tm and to the TnC subunit. The TnC subunit acts as a Ca2+ sensor in the muscle contraction process. When bound to Ca2+, TnC—which is also bound to TnI—causes TnI to shift on actin, which exposes the binding sites that allow myosin to bind. Despite the similar organization and function of the Tn complex in regulating skeletal and cardiac muscle contraction, some evidence suggests that there are subtle differences in the molecular interaction among Ca2+ and specific regions in the Tn isoforms. See Table 2 for key terms.
Key biology terms used in this article
Troponin-C
There are only 2 isoforms of TnC: cardiac troponin-C (cTnC) and fast twitch skeletal troponin-C (fsTnC). cTnC encodes for a 210 amino acid molecule with a molecular weight of 18 kDa that does not undergo alternative splicing.6 Structurally the cTnC and fsTnC protein subunits have similar C-terminal domains but have different N-terminal domains. There are 2 Ca2+ or magnesium ion (Mg2+) binding sites, Sites III and IV, on the C-terminal domain in both cTnC and fsTnC that play a role in anchoring TnC to other myofilaments within the cross-bridge. These high-affinity Ca2+ or Mg2+ binding sites help maintain structural integrity throughout the protein. It is the N-terminal domain of TnC that differs between the fast skeletal and cardiac isoforms of this protein. fsTnC contains 2 low-affinity Ca2+ binding sites (Sites I and II), which regulate muscle contraction. In cTnC, there is only one Ca2+ site (Site II), which acts as the regulatory region of cardiac muscle contraction. In cTnC, Site I does not bind Ca2+.7 To determine if the Ca2+ binding Site I could regulate cardiac muscle contraction, Sweeney et al8 mutated Ca2+ binding Site I to bind Ca2+ while also mutating Ca2+ binding Site II to no longer bind Ca2+. In this mutated muscle, cardiac muscle contraction was no longer triggered by Ca2+, confirming that Ca2+ binding Site II is the sole regulator of cardiac contraction.8 Ca2+ binding to TnC can also be regulated by other proteins in the cross-bridge, including interactions with TnI, TnT, actin, myosin, and Tm.1
Troponin-T
TnT contains 3 distinct genes that produce 3 different protein isoforms: (1) TNNT1 for slow twitch skeletal muscle (ssTnT), (2) TNNT2 for cardiac muscle (cTnT) and some neonatal skeletal muscles, and (3) TNNT3 for fast twitch skeletal muscle (fsTnT).9 The TnT protein isoforms undergo developmental, transcriptional, and translational regulation and are 220–300 amino acids long with a molecular weight of 30–35 kDa. The C-terminal domain and the middle portion of the protein bind to TnC, TnI, Tm, and Ca2+ and, therefore, directly interact with other proteins in the actin-myosin cross-bridge. The main difference among the 3 TnT protein isoforms is the N-terminal region, which plays a role in regulating conformational changes without directly binding to the cross-bridge. For example, in patients who undergo ischemia reperfusion injury, such as after a myocardial infarction (MI), there is a truncation of the N-terminal domain of cTnT by μ-calpain that alters cardiac contraction.10 Muscle fibers from transgenic mice overexpressing the truncated cTnT had an increased affinity for Tm, and ex vivo isolated working heart perfusions from these mice showed decreased ventricular contractile velocity but a preserved stroke volume.10 These data suggest that the posttranslational modification of the end of the N-terminal domain of cTnT during ischemia reperfusion is cardio-protective by improving/maintaining stroke volume in damaged hearts.
Further, cTnT is essential for development; studies conducted in rodents show that a complete loss of cTnT is embryonic lethal.11 There are 17 exons that are encoded by TNNT2, cTnT. Exons 4, 5, and 13 can undergo alternative splicing. Although the cause of exon 13 splicing is unknown, exon 13 plays a role in linking 2 functional domains of the TnT protein together.12 Alternative splicing of exon 5 is developmentally regulated in the heart; it is only expressed in the embryonic heart.13 This exon contains 10 amino acids that cause the protein to become more negatively charged; therefore, it binds Ca2+ more tightly, which decreases cardiac muscle contraction.13 Both embryonic and developing skeletal muscles also express cTnT that is alternatively spliced in a similar manner in the heart.14,15 This suggests that the alternative splicing of cTnT is regulated systemically and not in response to organ demands.
Exons 4 and 5 are alternatively spliced in the N-terminal variable region and give rise to 4 distinctive protein products: (1) cTnT1, no splicing; (2) cTnT2, splicing of exon 4; (3) cTnT3, splicing of exon 5; (4) cTnT4, splicing of exons 4 and 5 that have different molecular weights, Ca2+ sensitivity, and inhibition of force development.16 Both cTnT1 and cTnT2 have higher levels of Ca2+ sensitivity and are more likely to inhibit the ATPase activity of the actin-tropomyosin–activated myosin ATPase. The presence of exon 5 in the final cTnT protein product decreases the contractility of the actin-myosin cross-bridge. Furthermore, in various animal models of dilated cardiomyopathies, there are other alternatively spliced out exons in the cTnT protein that exacerbate disease. These models include exon 4 splicing in heart failure patients, familial hypertrophic cardiomyopathy, and diabetic rat hearts; exon 5 splicing in canine dilated cardiomyopathy; exon 6 splicing in abnormal cardiac structural changes in guinea pigs; exon 7 splicing in canine cardiomyopathy; and exon 8 splicing in dilated cardiomyopathy in turkeys.9 Further research is still needed to determine the functional significance of these alternatively spliced isoforms in human health and disease.
Troponin-I
TnI has 3 genes coding for 3 protein isoforms: (1) TNNI1 in slow twitch skeletal muscle (ssTnI), (2) TNNI2 in fast skeletal muscle (fsTnI), and (3) TNNI3 in cardiac muscle (cTnI). TnI interacts with both TnC and TnT and is able to inhibit muscle contraction by binding to actin with or without Ca2+; this region is termed the inhibitory peptide region.3,17 Ca2+ regulation of TnI is through TnI’s interaction with TnT and TnC but not direct binding of Ca2+ itself.9 TnI undergoes developmental and translational regulation and is 182–210 amino acids long with a molecular weight of 21–24 kDA.1 There are 8 exons that code for the various TnI genes; exons 4–8 are conserved among the 3 isoforms.18,19 cTnI is the largest of the 3 isoforms and contains a unique 30 amino acid N-terminal sequence coded by exons 1–3.19,20 There is no known alternative splicing that occurs in cTnI, but there are posttranslational modifications that affect muscle contraction.1,21 In cardiac myocytes, activation of protein kinase A (PKA) in response to β-adrenergic stimulation of the heart leads to phosphorylation of 2 adjacent serine residues: Ser-23 and Ser-24 in cTnI. These residues are part of a 32-residue extension of the N-terminal region of the protein that is unique to the cardiac isoform of TnI. Phosphorylation of these 2 serine residues leads to a decrease in affinity between cTnI and cTnC, a decrease in the sensitivity of cTnC to Ca2+, and an increase in the rate of relaxation.22 Interestingly, PKA-dependent phosphorylation of these specific serine residues in cTnI has decreased in the hearts of patients with heart failure.23,24
cTnI expression is developmentally regulated in the heart and is only expressed in the adult heart.25 This is important for clinical application of diagnostic tests, which use various Tn isoforms to determine cardiac damage. Protein and mRNA expression levels of the TnI isoforms were assessed during pre- and postnatal cardiac development and showed that ssTnI is expressed in the fetal and newborn heart; however, by 9 months postnatal, cTnI is the sole TnI fiber in the heart.26 The main difference between these 2 proteins is the cTnI contains a unique 27–33 amino acid N-terminal sequence. This sequence contains 2 phosphorylation sites that can further regulate TnI interactions with TnC and TnT through cyclic adenosine monophosphate protein kinase. Studies conducted in adult transgenic mice, containing ssTnI instead of wild-type cTnI in the heart, show no developmental dysfunction and no changes to mortality. The transgenic mice did exhibit diastolic dysfunction, which was exacerbated by treatment with the β-adrenoreceptor agonist isoprenaline. The ssTnI isoform has a lower affinity for TnC compared to TnI. The altered affinity changed the Ca2+ regulation in the transgenic mice and, therefore, caused cardiac dysfunction. Furthermore, recent studies have also shown that the C-terminal mobile domain of cTnI also plays a role in modifying cardiac contraction.17 Together, these data suggest that any mutations of cTnI can cause cardiac myopathies.
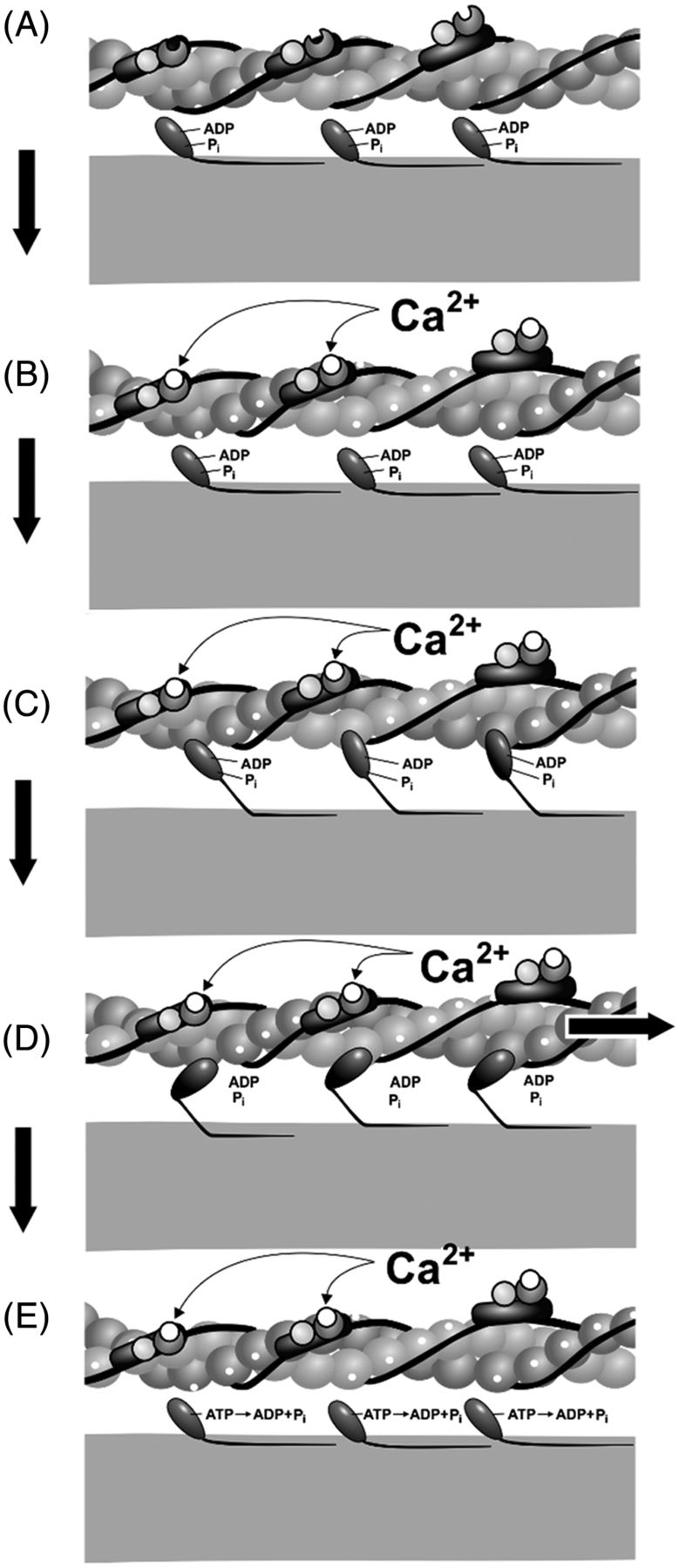
Both transcriptional and/or translational changes to the Tn subunits and isoforms can cause changes to muscle contraction through their interaction with the actin-myosin cross-bridge by altering the exposure of the binding sites on actin. After Ca2+ binds to Tn, the Tm shifts, which exposes the myosin-binding site on actin. The head of each myosin unit is now free to bind to the exposed binding site, forming a cross-bridge. Each myosin head has an adenosine diphosphate (ADP) and phosphate molecule attached to it from the previous cross-bridge cycle. Upon formation of the cross-bridge, the ADP and phosphate are released, which triggers a conformational change that results in the myosin head moving to the uncocked position. As the myosin head moves, it pulls the thin filament along with it (power stroke). Once the pivot occurs, the affinity for adenosine triphosphate (ATP) increases, and a new ATP attaches to the ATP-binding region in the myosin head. The myosin head then detaches from actin. ATP is then hydrolyzed to ADP and phosphate, and the cycle repeats itself until Ca2+ levels drop below the threshold or until maximal shortening of the sarcomere is achieved. Relaxation of the muscle cell occurs because of the action of ATP-dependent Ca2+ pumps moving Ca2+ back into the sarcoplasmic reticulum or out of the cell. As the cytoplasmic concentration of Ca2+ drops, the Tn-Tm complex slides back into place and blocks the myosin-binding sites on actin (Figure 2).

Power stroke image of muscle contraction. (A) Resting with myosin-binding sites covered, (B) calcium binding to troponin, myosin-binding sites exposed, (C) Cross-bridge formation as myosin binds to the myosin-binding sites on actin, (D) Power stroke which moves actin toward the M line, and (E) Dissociation of myosin head from actin. Image reprinted with permission of John Nagy.
Stimulation of cardiac muscle cells also leads to increased cytoplasmic concentrations of Ca2+. In these cells, action potentials lead to an influx of extracellular Ca2+ in addition to stimulation of Ca2+ release from the sarcoplasmic reticulum. Like skeletal muscle, the rise in intracellular Ca2+ levels lead to the removal of the Tm block by the interaction of Ca2+ and TnC. The role of Tn in muscle contraction is indispensable. The regulation of subunit isoforms is organ-specific during development or for diseases.
TROPONIN-CAUSING CARDIOMYOPATHIES
According to the National Institute of Health National Heart Lung and Blood Institute, cardiac myopathies are a group of diseases of the heart that are either inherited or acquired. A variety of hereditary cardiomyopathies—including hypertrophic cardiomyopathy (HCM), thickening of the heart muscle; restrictive cardiomyopathy (RCM), stiffness of the heart muscle; and dilated cardiomyopathy (DCM), thinning of the heart muscle wall—have been associated with mutations in the 3 subunits of cTn.6 A change as small as 1 amino acid in the Tn sequence, termed a point mutation, can cause changes to Tn subunit interactions in the actin-myosin cross-bridge formation, muscle contraction, and cardiac function. Over 100 mutations in cTn subunits—cTnT encoded by TNNT2, cTnI encoded by TNNI, and cTnC encoded by TNNC1—have been linked to cardiomyopathies.27
In 2001, the first HCM mutation in cTn was discovered in a 60-year–old male.28 To test the role of mutations found in cardiomyopathy patients, various in vivo and in vitro models have been studied. For example, the missense mutation, L29Q in TNNC1 was tested in a variety of in vivo and in vitro models, and results showed inconclusive effects on Ca2+ sensitivity.6 Other mutations—such as A8V, A31S, C84Y, and more—in cTnC cause changes to the Ca2+ sensitivity of Tn and, therefore, affect cardiac muscle contractility.6 Mutations in TNNC1 and TNNT2 have also been linked to DCM in sporadic and familial cases.29 These mutations affect the ability of Tn to bind to the actomyosin cross-bridge, altering Ca2+ sensitivity and muscle contraction. Lastly, mutations in the last 5 amino acids of the C-terminal domain of cTnI have been identified in various patients with HCM, DCM, and RCM.30 In vitro studies, cleaving the last 5 amino acids of cTnI, cause an increase in Ca2+ sensitivity of the Tn complex and thereby modify muscle contraction. These mutations are only some of the Tn mutations associated with cardiomyopathies, and other mutations are reviewed in Chang et al.31 Further studies, identifying other mutations in cTns in cardiomyopathies and the functional significance of these mutations, will play an important role in treating patients in the future.
DEGRADATION OF TROPONIN IN TISSUE AND RELEASE INTO SERUM
The Tn complex is a tightly regulated protein that is essential for striated muscle contraction. However, its importance in detecting ischemia-related cardiac muscle injuries is recognized, especially with the appearance of the high-sensitivity assays based on cTnI and cTnT. To maintain homeostasis, a functional heart is needed to deliver nutrients and oxygen to different body parts as well as itself. About 4%–5% of the cardiac output goes to the heart for supplying its pumping activities.32 When cardiac myocytes die or are injured, Tn is released from these cells and diffuses into the circulation. The release of Tn from cardiac myocytes is mediated through the processes of inflammation, apoptosis, or necrosis. It is worthy to note that very low concentrations of cardiac biomarkers can be detected in people with healthy hearts by the recently developed high-sensitivity cTn assay.33,34 Aside from troponins, other biomarkers are released into circulation when myocardial injury occurs, including creatine kinase, lactate dehydrogenase, and myoglobin, to name a few.35 The time for these various biomarkers to diffuse from myocardial cells into the circulation is determined by the earliest time when they could be detected from blood samples after myocardial incidents. Together with the presenting clinical symptoms, measuring these biomarkers help clinicians diagnose cardiac diseases more accurately.
Cardiac troponins are released into the bloodstream 2–8 hours after a cardiac injury occurs, peak at 12–48 hours, and can remain at detectable levels for up to 3–5 days for cTnI and 5–10 days for cTnT. Therefore, there is a natural rise and fall pattern associated with biomarkers after acute myocardial injury. The concentration of cTnI or cTnT is positively related to the number of injured cardiac myocytes (area of ischemia/necrosis).36 Furthermore, the added value of troponins in the diagnosis of myocardial injury ensures that the level of Tn remains elevated up to 7–10 days after acute MI.36 In regards to what was previously discussed in this article, because there is no cardiac-specific TnC, TnC is not used as a clinical diagnostic marker. Unfortunately, in addition to myocardial injuries, cardiac damage from other diseases—such as kidney disease, sepsis, and severe anemia—can also cause an elevation in Tn. For example, patients with end-stage renal disease have increased cTnI and cTnT concentrations, and these biomarkers are prognostic for the adverse events of the end-stage renal disease.37 These diagnostic markers are used differently for different prognoses and diagnoses. While both cTnT and cTnI levels are usually measured, cTnI was shown to be the preferred biomarker for myocardial damage in patients with chronic renal failure.38
TROPONIN AS A DIAGNOSTIC AND PROGNOSTIC BIOMARKER
It is a challenge to accurately diagnose patients presenting with symptoms of acute coronary syndrome, especially for those who do not exhibit the classic changes in ST-segment of electrocardiogram recordings. As a result, cardiac markers have played an increasingly important role in aiding in the diagnosis of acute MI, especially within the last 2 decades. Troponins provide valuable information for helping establish a diagnosis in the case of chest pain presentation. cTnI or cTnT have been recognized as the preferred biomarkers for detecting MI since the Joint European Society of Cardiology and the American College of Cardiology published a definition for MI in 2000.39 As myocytes die, they sequentially release intracellular components into the blood that are used as biomarkers, which can be detected at specific times after injury by different types of immunochemical assays. Various developed assays for detecting cTnI and cTnT are used worldwide and are key biomarkers of MI in patients.36,40 Although elevated cTnI is an indicator of MI, patients with other chronic diseases also express elevated TnI levels in their blood. For example, patients with Fabry disease have an increased cTnI, especially for those with a left ventricular hypertrophy.41 Therefore, when examining blood levels of Tn, it is important to incorporate patient history and symptoms to make an accurate diagnosis.
cTn has been shown to have prognostic value as well.42,43 The cTnT can be used for prognostic purposes for nonemergent-postpercutaneous coronary intervention outcome.44 In a prospective cohort study of 1,024 patients with unstable angina/non-ST–segment elevation MI, who underwent coronary angiography and subsequent coronary stenting within 24 hours, the risk of in-hospital and long-term mortality was greater with increased levels of baseline cTnT.42 These data indicate that cTnT is a useful biomarker for post MI prognosis purpose.
Although the current diagnostic systems can reliably diagnose MI 3–6 hours after the onset of MI, efforts have been made to detect changes in Tn biomarkers earlier with the use of high-sensitivity assays.39,45⇓-47 High-sensitivity assays have been developed, are available from both Abbott and Roche, and will be discussed more fully in the second and third articles in this focus series. The use of these high-sensitivity assays is going to be evaluated from sensitivity and specificity perspectives for many cardiac diseases, including MI. Undoubtedly, a thorough knowledge on Tn biomarkers is critical for health care providers to either diagnose or make prognoses in coronary artery or heart diseases.
- Received July 11, 2018.
- Accepted November 27, 2018.
American Society for Clinical Laboratory Science
References




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Keywords
- ADP - adenosine diphosphate
- ATP - adenosine triphosphate
- ATPase - adenosine triphosphatase
- Ca2+ - calcium
- cTn - cardiac troponin
- cTnC - cardiac troponin-C
- cTnI - cardiac troponin-I
- cTnT - cardiac troponin-T
- DCM - dilated cardiomyopathy
- fsTnC - fast twitch skeletal troponin-C
- fsTnI - fast twitch skeletal troponin-I
- fsTnT - fast twitch troponin-T
- HCM - hypertrophic cardiomyopathy
- Mg2+ - magnesium ion
- MI - myocardial infarction
- mRNA - messenger RNA
- PKA - protein kinase A
- RCM - restrictive cardiomyopathy
- ssTnI - slow twitch skeletal troponin-I
- ssTnT - slow twitch skeletal troponin-T
- Tm - tropomyosin
- Tn - troponin
- TnC - troponin-C
- TnI - troponin-I
- TnT - troponin-T
- troponin
- high-sensitivity troponin assays
- myocardial infarction